Proces naukowy, prowadzący ostatecznie do odsłonięcia kolejnych tajemnic natury, jest często długotrwały i przypomina błądzenie po omacku w gęstej mgle. Zza zasłony niewiedzy co i rusz wyłaniają się zarysy rzeczy, o bardzo obiecujących kształtach, które po bliższym zapoznaniu nie muszą okazać się tym czego szukano. Zwykle zatem badacz dociera do odkrycia metodą prób i błędów. Jednak obraz nauki, jaki przedstawiają często podręczniki i media, obejmuje zazwyczaj wyłącznie sukcesy, udane doświadczenia i potwierdzone hipotezy. Stąd też dziwne się może wydać przeciętnemu odbiorcy, że taki na przykład Mendelejew wiele lat pracował nad ułożeniem swej tabeli pierwiastków, albo że nawet dwa wieki po zbudowaniu teleskopu wątpiono w to, że Ziemia krąży wokół słońca i obraca się sama wokoło swej osi.
Te osobliwości stają się łatwiejsze do zrozumienia, jeśli uświadomić sobie, że historia nauki – tak zresztą jak i historia cywilizacji – jest historią zarówno odkryć jak i pomyłek, zwycięstw i porażek, zaś to jakie błędy popełniano dawniej, jest zarówno świadectwem czasów w jakich się zrodziły, jak i przestrogą dla nas współczesnych, abyśmy z większą ostrożnością podchodzili do naszych dzisiejszych osądów i dokonań W tej prezentacji zajmę się błędami i wypaczeniami w historii Chemii, zawężając temat do tych niepowodzeń, jakie dotyczyły odkryć pierwiastków chemicznych. Pozwoliłem sobie podzielić wyszukane przypadki na trzy zasadnicze grupy, choć mój wybór tego, który do której ma być zaliczony, jest dosyć arbitralny. Pierwszą taka grupą będą zatem:
1. Substancje idealne.
Do podstawowych mechanizmów procesu naukowego, należy formułowanie hipotez, stawianie tez i ich doświadczalna weryfikacja, zanim jednak następuje ten ostatni etap, częstokroć przez długi czas funkcjonują pewne hipotetyczne pojęcia, zaś opisywane nimi twory uznawane bywają za prawdziwe – dotyczy to też substancji, których istnienie tłumaczyć miało wedle danej teorii, obserwowane zjawiska.
 |
| Siedem elementów pierwotnych na siedemnastowiecznej alchemicznej rycinie |
Jednym z najwcześniejszych takich przypadków, był powstały w starożytnej Grecji pogląd, że wszystkie substancje materialne składają się z pewnej ograniczonej liczby podstawowych elementów, których różne połączenia stanowią materię rzeczy – pogląd nie aż tak odległy od współczesnego – przy czym mający największy wśród filozofów autorytet
Arystoteles, ograniczył liczbę tych pierwiastków do czterech, kojarząc je z
czterema żywiołami: Ogniem, Powietrzem, Wodą i Ziemią. Ich obecność i proporcje w substancjach, decydowały o ich własnościach.
W późniejszy okresie alchemicy próbowali wpływać na zawartość żywiołów w materii, sądząc iż uda się tym sposobem przemienić jedne w inne, na przykład pospolity ołów w cenne złoto.
Właściwie dopiero w czasach nowożytnych pogląd ten został podważony, i zastąpiony pojęciem pierwiastków chemicznych, równocześnie jednak pojawiła się inna teoria; mianowicie niemiecki chemik J. Becher uznał ogień za zjawisko, nie zaś żywioł; równocześnie jednak założył, że istnieje pewien specjalny pierwiastek palności –
Flogiston - uwalniany z płomieniem. Po oddzieleniu flogistonu z substancji powstawał popiół, będący miałkim, lżejszym od pierwotnej materii pyłem. Jednak wraz z rozwojem badań pojawiało się coraz więcej obserwacji, których nie dawało się w tak prosty sposób wytłumaczyć.
Sprawą palności i płomienia zajął się w XVII wieku markiz
Lavoisier, stwierdzając doświadczalnie rolę tlenu w tym procesie i właściwie interpretując przemiany ciał, jednak i w jego prace wkradł się błąd. Lavoisier był przekonany o konieczności obecności tlenu w kwasach mineralnych, co znalazło odzwierciedlenie w jego łacińskiej nazwie „Oxygenium” – czyli „Kwasoród” – jak to próbował tłumaczyć Jędrzej Śniadecki. W tym kontekście problem stwarzał kwas solny, który można było otrzymać łącząc chlor z wodorem, a zatem najwyraźniej nie zawierał on tlenu. Autorytet francuskiego chemika był tak wielki, że już odkrywca uznał chlor za bardzo trwały tlenek nie wyodrębnionego pierwiastka, nazwanego „
muriaticum” – stąd spotykana niekiedy stara łacińska nazwa kwasu solnego „acidum muriaticum”. Dopiero odkrycia jodu i bromu, oraz niemożność rozłożenia chloru znanymi sposobami, przekonały naukowców o błędności teorii.
W 1815 roku William Prout zaproponował teorię budowy atomów, w pewnym stopniu podobną do koncepcji dawnych filozofów. Uznał mianowicie, że atomy innych pierwiastków, to w rzeczywistości cząsteczki składające się z różnej ilości atomów jedynego prawdziwego pierwiastka, wodoru, który przy okazji przemianował na
protyl Masy wszystkich pierwiastków miały być więc wielokrotnościami mas wodoru. Jednak dokładniejsze badania obaliły ten pomysł. Na przykład zmierzona masa atomu chloru wynosiła 35,5 a to stanowiło niecałkowitą wielokrotność.
Hipoteza ta miała jednak w sobie pewien genialny koncept, który w pewnej odmianie przyjął się później. Zakładała mianowicie, że atomy składają się z pewnej ilości podstawowych cząstek, których liczba wywołuje określoną masę atomu. Jednostkami tymi są nukleony, w tym protony i neutrony. Masa atomowa chloru wynika z obecności dwóch izotopów, różniących się ilością neutronów, chloru-35 i chloru-37.
W XIX wieku, nowe substancje „tworzył” trend ówczesnej fizyki, aby znane oddziaływania tłumaczyć mechanicznie, obecnością specyficznych substancji. Tak więc światło rozchodzić się miało w substancji zwanej
Eterem, zaś przekazywanie ciepła być przepływem
Cieplika. Zarówno Maxwell jak i Joule, tworząc swe poprawne w obserwacjach teorie fizyczne, myśleli o opisie zachowań tych nieważkich, przenikliwych, niewidzialnych cieczy.
2. Pierwiastki niedoszłe.
Rozwój technik analitycznych w dawnych badacza ciekawość, pchającą ich do poddawania badaniom wszystkiego co wpadło im w rękę. Wówczas też zaczął się prawdziwy wysyp nowych pierwiastków – czasem jednak dochodziło do mniej lub bardziej spektakularnych pomyłek. Pomyłki zdarzały się najlepszym. Młody
Berzelius badając szwedzkie minerały, doniósł kiedyś o odkryciu nowego pierwiastka, który w nawiązaniu do skandynawskiej mitologii nazwał
Torem. Przesłał próbki pierwiastka innym badaczom, a ci orzekli jednogłośnie, że jest to pospolity fosforan itru, wobec czego młody chemik z zawstydzeniem wycofał odkrycie. Kiedy znacznie później, już jako znany chemik, ponownie wyodrębnił nieznany pierwiastek z jednego z minerałów, użył starej nazwy, tym razem szczęśliwie.
Tor przyniósł zresztą podobny problem innemu chemikowi, Charlesowi Baskervillowi, który w 1901 roku ogłosił, że wyodrębnił z niego dwa nowe pierwiastki. Jeden dla uhonorowania poprzednika nazwał
berzelium, a drugi dla upamiętnienia amerykańskiego stanu w którym pracował, nazwał
carolinium. Obie wyodrębnione frakcje były jednak mieszankami toru z innymi znanymi pierwiastkami.
Gdy
Kirchhoff i B
unsen odkryli spektroskopowo kilka metali alkalicznych w wodach mineralnych, a
Janssen hel w koronie Słońca, w widmie mgławicy emisyjnej Kocie Oko wykryto nieznaną linię widmową. Uznano, że świadczy ona o obecności nowego pierwiastka, nazwanego
nebulium. Podobna rzecz stała się gdy badano widmo zorzy polarnej – tam zielona linia emisyjna wskazywała na obecność
auorium. Natomiast w widmie korony słonecznej wykryto linię
coronium. Dopiero gdy lepiej poznano zjawiska emisji i wzbudzenia, stwierdzono że nebulium i aurorium to różnie zjonizowany tlen. Natomiast coronium okazało się linią widmową wielokrotnie zjonizowanego żelaza, dającego się zapisać jako Fe
13+
Dużo kłopotu sprawiły chemikom „pierwiastki ziem rzadkich”, czyli
Lantanowce, a to z uwagi na bardzo duże podobieństwo właściwości. Prawie wszystkie występują na raz w jednym minerale – Monacycie – a oddzielenie jednego od drugiego aż do wynalezienia chromatografii jonowymiennej nastręczało duże trudności. Wtedy też wyodrębniono „ziemię”
didimium, przez długi czas uważaną za pierwiastek, zanim nie rozdzielono jej na kilka osobnych pierwiastków. Śladem jej istnienia są nazwy dwóch lantanowców: Neodymu i Prazeodymu.
W 1925 roku radziecki chemik Dobroserdow wykrył słabą radioaktywność
próbek soli potasu. Uznał więc, że może to być sygnał pochodzący od
przewidywanego najcięższego pierwiastka alkalicznego, leżącego pod
Cezem, i nazwał go
Russium od nazwy swego kraju. Później okazało
się jednak, że naturalny trwały potas zawiera domieszkę radioaktywnego
izotopu K-40. Najcięższy litowiec sprawił problemy także innych
badaczom. Badania rentgenowskie siarczanu magnezu doprowadziły dwóch
angielskich chemików do ogłoszenia odkrycia
alkalinium, później Fred Allison badając spektroskopią efekty opto-magnetyczne miał wykryć w minerałach litu pierwiastek
virginium, wreszcie w latach 30. rumuńska fizyczka miała badaniami rentgenowskimi wykryć w tychże minerałach
moldavium.
Wszystkie te doniesienia okazały się oczywiście błędne, bowiem
poszukiwany pierwiastek frans jest radioaktywny i ma tak krótki czas
półtrwania, że nie występuje w minerałach.
W miarę upływu lat zastanawiającą sprawą była luka w układzie okresowym, obejmująca pierwiastek nr. 43. Wszystkie okoliczne pierwiastki były już znane, i występowały w przyrodzie, natomiast tego jednego nie dawało się odnaleźć. Wreszcie w 1925 roku Walter Noddack i jego żona Ida Tacke postanowili otrzymać go sztucznie. Bombardowali neutronami minerał kolumbit - jakże nieszczęsny dla odkrywców - a podczas analizy wykryli w nim poszukiwany pierwiastek, nazwany
Masurium dla uczczenia Mazur - regionu skąd pochodzili. Odkrycie nie zostało jednak uznane i trzeba było czekać aż do 1937 roku, kiedy to otrzymano go bombardując neutronami molibden i otrzymując
Technet. Obecnie jednak uznaje się za prawdopodobne, że odkrycie wcześniejszego zespołu mogło być prawdziwe.
Trzecią grupą przypadków są:
3. Pierwiastki zapoznane
Przypadki odkrywania czegoś więcej niż raz nie są tak rzadkie w historii; przykładem choćby druk, wynaleziony w Chinach, później w Europie, czy pomysł budowania piramid, wynaleziony w tak odizolowanych miejscach globu jak Egipt i Ameryka Środkowa. W przypadku Chemii sytuacja taka możliwa jest wtedy, gdy albo odkrycie nie zostanie potwierdzone z braku odpowiednich metod analitycznych, albo z innych, nieraz losowych, nie przewidzianych okoliczności.
W
1801 roku profesor
Andres Manuel del Rio odkrył w Meksyku minerał, z którego wydzielił nieznany pierwiastek o barwnych związkach, który dla czerwonej barwy jednego z tlenków nazwał
erythronium . Profesor wysłał próbki do Europy, aby tam potwierdzono jego przypuszczenia, lecz pierwszych kilka zaginęło, zaś pierwsza dokładna analiza, sugerująca obecność w próbkach nieznanego pierwiastka, zatonęła wraz ze statkiem którym ją przesyłano. Dysponując tylko wstępną analizą, sugerującą, że próbki zawierają chrom, del Rio wycofał zgłoszenie. Dopiero w 1831 roku Szwed Nils Gabriel Sefström odkrył barwny pierwiastek w rudach żelaza, nazywając go
Wanadem od skandynawskiej bogini Vanadis (Freya). Okazał się on występować w meksykańskim minerale badanym przez del Rio, nazwanym potem wanadynitem
.jpg/1024px-Vanadinite%2C_goethite(2).jpg) |
| Wanadynit w rudach żelaza |
W tymże, pechowym najwyraźniej,
1801 roku, mineralog
Hatchett wyodrębnił w południowoamerykańskiego minerału
kolumbitu, nieznany metal podobny do
Tantalu, który nazwał
kolumbem. Jednak nie wszystkie analizy potwierdziły jego wyniki, a w dodatku inny mineralog Wollason udowodnił, że kolumbit jest tym samym minerałem co tantalit. Uznano zatem, że Kolumb to Tantal, a Hatchett się pomylił.
Jednak w 1848 roku Henrie Rose wyodrębnił z kolumbitu nowy pierwiastek podobny, ale różny od tantalu. Nie wiedząc o dawnym odkryciu, które uznane za pomyłkę zesłano do lamusa, nazwał swój pierwiastek
Niobem, nawiązując do mitologii greckiej (
Niobe była córką
Tantala). Naukowcy z ameryki południowej próbowali później odkręcić powstałe zamieszanie, ale nowa nazwa się przyjęła. Można by żartobliwie zauważyć, że odkrycia nowych pierwiastków odsuwały się od tamtego kontynentu, niczym woda i jadło od Tantala.
 |
| Krystaliczny niob |
Podobny przypadek miał miejsce później, podczas poszukiwań ostatniego z lantanowców. Gdy
Welsbach wydzielił z tlenku itru pierwiastek nazwany
Kasjopem, w innej części Europy Urban odkrył
Lutet. Sprawa pierwszeństwa i nazwy nowego pierwiastka, stała się sprawą polityczną – Welsbach był Niemcem, a Urban Francuzem. Oba kraje od dawna rywalizowały we wszystkich dziedzinach, czego niechlubnym przykładem historia „
promieni N” będących francuską odpowiedzią na „
promienie X”, które okazały się ciekawym przykładem zbiorowej sugestii. Ostatecznie komisja naukowa uznała pierwszeństwo Urbana.*
Dla nas najciekawsze są dwa ostatnie przypadki, wiążą się bowiem z naszym krajem. W
1808 roku, świetny polski chemik
Jędrzej Śniadecki, ogłosił rozprawę na temat nowego pierwiastka, odkrytego w pozostałościach po rozpuszczeniu platyny, który od nowo odkrytej planety (dziś uznawanej za planetoidę) nazwał
Westem. Pisma na ten temat publikował głównie w języku polskim, wysłał też sprawozdanie do francuskiej akademii nauk. Niestety Francuzi nie potwierdzili odkrycia, nie stwierdzając aby w platynie zawarty był jeszcze jeden pierwiastek.
Dopiero w 1848 roku, Claus wykrył w syberyjskiej platynie pierwiastek, nazwany
Rutenem, od dawnej nazwy Rusi. Niestety odkrycia Śniadeckiego nie w sposób zweryfikować, bo ostatnie próbki Westu zaginęły po śmierci naukowca.
Ostatni przypadek dotyczy pospolitego pierwiastka
Tlenu. Do dziś trwają spory co do tego kto jest jego pierwszym odkrywcą. Jako pierwiastek opisał go Lavoisier,
Scheele opisał gaz wydzielający się z rozkładu tlenku rtęci, zaś
Priestley gaz powstający z rozkładu azotanu potasu. Być może podobne obserwacje czyniło przed nimi wielu badaczy, wiadomo jednak że polski alchemik
Sędziwój, opisał w XVI wieku gaz, powstający z rozkładu saletry, więc w pewnym sensie był pierwszym który opisał tlen, nie wiedząc, że jest to nowy, nieznany pierwiastek.
Tak więc nie z samych sukcesów składa się historia Chemii, a błędy, niedokładności, oszustwa a często i zwykłe przypadki, przeszkadzały w dokonaniu odkrycia. Może jednak na błędach, które dawniej popełniali inni, my współcześni nauczymy się, jak samemu się przed nimi ustrzec.
---------
* bezstronność tej komisji jest jednak wątpliwa - składała się z czterech członków a jednym z nich był Urban
Korzystałem głównie ze stron anglojęzycznych, przeważnie z haseł Wikipedii dotyczących wymienianych pierwiastków, oraz z Księgi Pierwiastków Chemicznych, która naprowadziła mnie na pomysł, mogę jednak polecić następujące strony:
* Misidentified_chemical_elements
*
The curious case of Columbium
*
http://sciagawa.com.pl/Chemia/Chemia-inne/Jedrzej-Sniadecki-wielki-uczony/6
*
http://ciekawe.onet.pl/przyroda/vestium-vel-ruten,1,4405067,artykul.html
 Kryształ fuksyny w otoczeniu gronkowców gramm+. Zrobione przy okazji wybarwiania preparatu mikrobiologicznego metodą Gramma na zajęciach bioanalizy jeszcze w technikum.
Kryształ fuksyny w otoczeniu gronkowców gramm+. Zrobione przy okazji wybarwiania preparatu mikrobiologicznego metodą Gramma na zajęciach bioanalizy jeszcze w technikum.





 W ramach pracowni analizy związków organicznych. Zrobiłem też filmik stapiania związku z metalicznym sodem, ale ponieważ jedną ręką trzymałem szczypce a drugą aparat, z braku koordynacji nie sfilmowałem momentu rozbijania próbówki w tygielku z wodą.
W ramach pracowni analizy związków organicznych. Zrobiłem też filmik stapiania związku z metalicznym sodem, ale ponieważ jedną ręką trzymałem szczypce a drugą aparat, z braku koordynacji nie sfilmowałem momentu rozbijania próbówki w tygielku z wodą.
 Wzór przypomina trochę kolonię pleśni lub dendryt.
Wzór przypomina trochę kolonię pleśni lub dendryt. Oczywiście wiedziałem co to jest, ale przypomnienie sobie tego jednego faktu trącało w pamięci inne, w pewnym stopniu powiązane z tym że dziś prowadzę tego bloga, to zaś nasunęło mi na myśl pomysł ciekawej notki w sam raz tu pasującej. Ale zanim opowiem jak to w mojej przeszłości z tą Chemią było, zacznę ab iowi albo wręcz ab mala* to jest od narośli:
Oczywiście wiedziałem co to jest, ale przypomnienie sobie tego jednego faktu trącało w pamięci inne, w pewnym stopniu powiązane z tym że dziś prowadzę tego bloga, to zaś nasunęło mi na myśl pomysł ciekawej notki w sam raz tu pasującej. Ale zanim opowiem jak to w mojej przeszłości z tą Chemią było, zacznę ab iowi albo wręcz ab mala* to jest od narośli: Zaczyna się od tego, że latem mały owad podobny do meszki, nazywany Galasówką Dębianką (Cynips quercus-folii), nacina listek dębu i składa w nacięciu jajeczko. Równocześnie wstrzykuje do liścia pewne wydzieliny, skłaniające tkankę twórczą liścia (kallas) do tworzenia torbieli otaczającej jajeczko. Z jajeczka powstaje larwa która również wydziela podobną substancję. Wokół rozwijającej się larwy rozrasta się kulista narośl z miękkiej, gąbczastej tkanki poprzecinanej licznymi żyłkami doprowadzającymi odżywcze soki, z odłożoną pożywną skrobią i białkiem. Larwa tkwi tam niczym zarodek w jajku, wraz z listkiem opada, zimuje, a wczesną wiosną wykluwa się do postaci dojrzałej.
Zaczyna się od tego, że latem mały owad podobny do meszki, nazywany Galasówką Dębianką (Cynips quercus-folii), nacina listek dębu i składa w nacięciu jajeczko. Równocześnie wstrzykuje do liścia pewne wydzieliny, skłaniające tkankę twórczą liścia (kallas) do tworzenia torbieli otaczającej jajeczko. Z jajeczka powstaje larwa która również wydziela podobną substancję. Wokół rozwijającej się larwy rozrasta się kulista narośl z miękkiej, gąbczastej tkanki poprzecinanej licznymi żyłkami doprowadzającymi odżywcze soki, z odłożoną pożywną skrobią i białkiem. Larwa tkwi tam niczym zarodek w jajku, wraz z listkiem opada, zimuje, a wczesną wiosną wykluwa się do postaci dojrzałej. . Podczas gdy tam obok grupy karboksylowej, w położeniu orto znajdowała się jedna grupa hydroksylowa, przez co związek był właściwie pochodną fenolu, tutaj mamy aż trzy takie grupy, dwie położone w pozycjach meta i jedna w para.
. Podczas gdy tam obok grupy karboksylowej, w położeniu orto znajdowała się jedna grupa hydroksylowa, przez co związek był właściwie pochodną fenolu, tutaj mamy aż trzy takie grupy, dwie położone w pozycjach meta i jedna w para. Związek ten jest używany w histologii, gdyż trwale zabarwia jądro komórki, pozwalając dobrze zbadać jego właściwości. Ponieważ jednak jego podaż z naturalnych źródeł skurczył się wraz z lasami deszczowymi w których rośnie kampeszyn, w ostatnich latach zaczęto używać w zastępstwo innych barwników. Nazwa zwyczajowa drzewa wzięła się zapewne stąd, że drewno wskutek powstawania omówionego kompleksu zabarwiało się niebieskawo, podobnie jak drewno olchy przebarwia się na czerwono.
Związek ten jest używany w histologii, gdyż trwale zabarwia jądro komórki, pozwalając dobrze zbadać jego właściwości. Ponieważ jednak jego podaż z naturalnych źródeł skurczył się wraz z lasami deszczowymi w których rośnie kampeszyn, w ostatnich latach zaczęto używać w zastępstwo innych barwników. Nazwa zwyczajowa drzewa wzięła się zapewne stąd, że drewno wskutek powstawania omówionego kompleksu zabarwiało się niebieskawo, podobnie jak drewno olchy przebarwia się na czerwono. Ten prosty w wykonaniu atrament ma też swoją ciemną stronę - przyczynia się do degradacji papieru i pergaminu, zwłaszcza w przypadku bardzo starych manuskryptów. Z czasem kwas galusowy zawarty w artamencie utleniał się i blakł do koloru żółtego lub pomarańczowego, pozostawiając pewną ilość wolnych jonów żelaza III, te reagowały z podłożem dając rdzawe obwódki wokół liter. Gdy papier lub pergamin utleniał się w wyniku dostępu powietrza i światła, część uwolnionych elektronów redukowała Fe3+ do Fe2+, te jony chętnie z powrotem się utleniają, redukując materiał podłoża, a utlenione jory redukują się znów po pewnym czasie...
Ten prosty w wykonaniu atrament ma też swoją ciemną stronę - przyczynia się do degradacji papieru i pergaminu, zwłaszcza w przypadku bardzo starych manuskryptów. Z czasem kwas galusowy zawarty w artamencie utleniał się i blakł do koloru żółtego lub pomarańczowego, pozostawiając pewną ilość wolnych jonów żelaza III, te reagowały z podłożem dając rdzawe obwódki wokół liter. Gdy papier lub pergamin utleniał się w wyniku dostępu powietrza i światła, część uwolnionych elektronów redukowała Fe3+ do Fe2+, te jony chętnie z powrotem się utleniają, redukując materiał podłoża, a utlenione jory redukują się znów po pewnym czasie...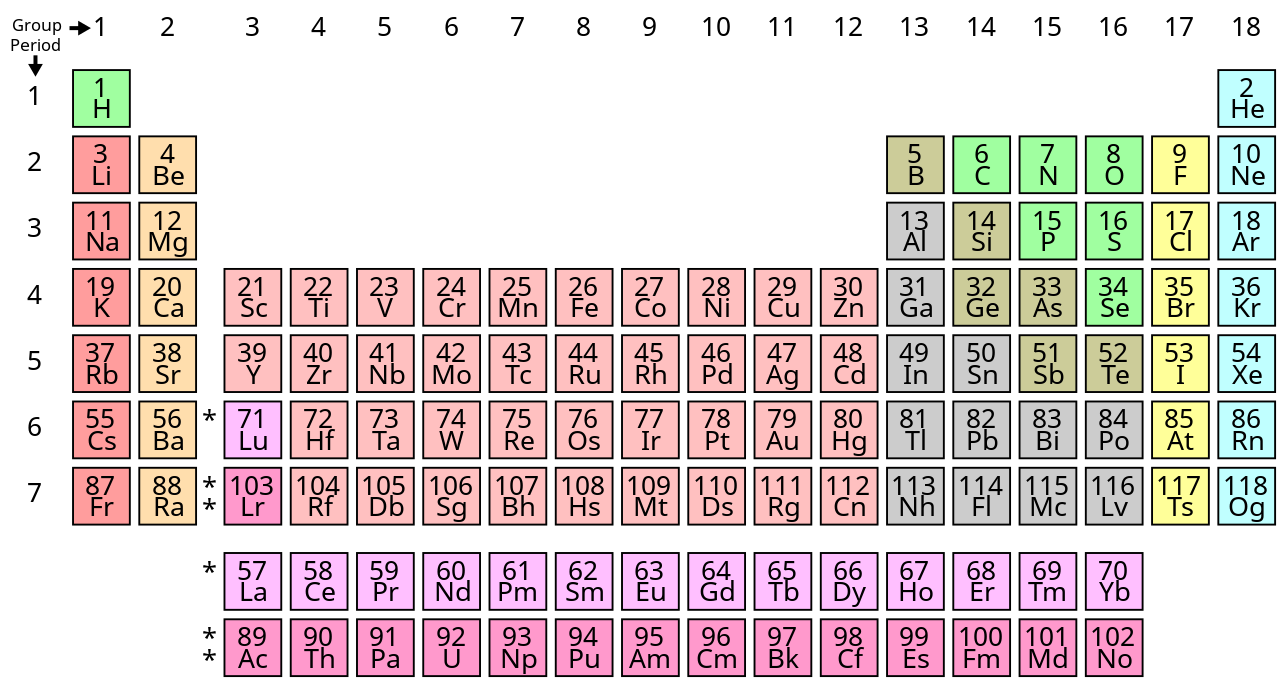

{kind=link}