Mieszadła magnetyczne to obecnie jeden z najpopularniejszych sprzętów laboratoryjnych. W podstawce, często podgrzewanej, znajdują się magnesy, które działają na magnetyczny "drops" wrzucany do naczynia, pozwalając na wygodne i szybkie mieszanie. Magnes wkładany do naczynia zwykle jest chroniony warstwą białego teflonu, materiału na tyle niereaktywnego, że wytrzymuje mieszanie nim kwasów i środków utleniających.
Jak wynika z niedawnej publikacji badaczy z Rice University, teflon w takich mieszadłach czasem jest jednak niedostatecznie niereaktywny.

Prowadzili oni dodawanie grup funkcyjnych na powierzchni nanorurek, stosując jako jeden z etapów redukcję Billupsa-Bircha. To modyfikacja znanej redukcji metalicznym sodem w ciekłym amoniaku, w której następuje równoczesna alkilacja powierzchni węglowej w miejscach defektów sieci. W tym konkretnym przypadku funkcjonalizowano nanorurki borazynowe, a więc ze związku złożonego z atomów boru i azotu w stosunku 1:1. O dziwo w czasie reakcji zawiesina kremowych rurek stała się szara, natomiast magnetyczne mieszadełko stało się czarne. Standardowo używana do takich cząsteczek analiza termograwitacyjna nie wykazała, aby z rurkami stało się coś złego. Jednak nietypowy kolor był wyraźnie widoczny. Problemem było też uzyskanie spójnych wyników, które raz wskazywały na wysoki stopień podstawienia a kiedy indziej na bardzo niski.
Dokładniejsze badania wykazały, że w warunkach reakcji lit rozpuszczony w amoniaku reaguje z teflonem. Następuje to w szybkiej reakcji rodnikowej, która przeszkadza w alkilowaniu rurek, w dodatku podstawiając je dodatkowymi, nie planowanymi grupami. Dlatego te otrzymywane przy pomocy świeżych mieszadełek były słabo podstawione, a te ze starymi, już poczernionymi, reagowały lepiej. Ponieważ dotychczas używano tej reakcji do modyfikacji rurek węglowych, które są czarne, naukowcy sądzili w takich przypadkach, że zmiana koloru magnesów wynika z osadzania się rurek na teflonie. A w każdym razie, że nawet jeśli teflon się zmieniał to nie musiało to wpływać na wyniki.
Gdy badacze zamienili mieszadełka na takie o szklanej otoczce, wyniki stały się powtarzalne, a nanorurki nie ciemniały. [a]
Sam obserwowałem trwałe ciemnienie mieszadełek po reakcjach ze środkami alkilującymi i chlorującymi, ale nie stwierdziłem aby wpływało to na wydajność czy powstawanie ubocznych produktów.
Wodorek helu wreszcie wykryty
Gdy po Wielkim Wybuchu wszechświat się rozrzedzał i chłodził, początkowa plazma różnych nietrwałych, naładowanych cząsteczek zaczęła się stopniowo łączyć w obojętne cząstki. To wtedy przestrzeń nabrała dostatecznej przezroczystości dla światła, aby dało się coś zobaczyć. Najwięcej powstawało atomów wodoru, cząsteczek wodoru i atomów helu, może z domieszką litu. Wśród powstających molekuł powinien się pojawić też kation wodorku helu HeH+. Jest to nietrwałe połączenie, proton łatwo dysocjuje (pKa=63), dlatego cząsteczka istnieje bądź jako element równowagi między helem i wodorem w zagęszczonych, zjonizowanych gazach, lub jako składnik gazów tak bardzo schłodzonych i rozrzedzonych, że w trakcie swego trwania nie ma z czym zareagować.
Obserwowano ją w eksperymentach na ziemi, ale w kosmosie o dziwo nie udawało się jej wykryć. Aż do teraz. W niedawnej publikacji badaczy z Instytutu Radioastronomii Maxa Plancka potwierdzono istnienie charakterystycznej linii emisyjnej 149.1 µm, której wykrycie dotychczas uniemożliwiała słaba rozdzielność spektralna i przeszkadzające zanieczyszczenia w atmosferze. Bardzo blisko tej linii znajduje się jeden z sygnałów wiązania C-H (149.09 µm) w związku z czym proste węglowodory obecne w przestrzeni mogą maskować szukaną linię, w dodatku w zakres ten częściowo wchodzi pochłanianie przez wodę w atmosferze, co dodatkowo osłabia sygnały. Badacze skorzystali więc z udostępnionego przez NASA teleskopu na pokładzie dużego samolotu, który z wysokości 12 kilometrów przeprowadził pomiar widma w mgławicy NGC 7027. Sygnały były już dostatecznie mocne, aby możliwe było odfiltrowanie dwóch bliskich linii widmowych i potwierdzenie istnienia szukanej molekuły.[b]
Rekordowy poziom dwutlenku węgla
Średnia globalna zawartość dwutlenku węgla atmosferze przekroczyła w kwietniu 410 ppm, z kolei w obserwatorium na Mauna Loa przekroczyła 415 ppm. Teraz zapewne średnie miesięczne zaczną spadać zgodnie z rocznym cyklem:
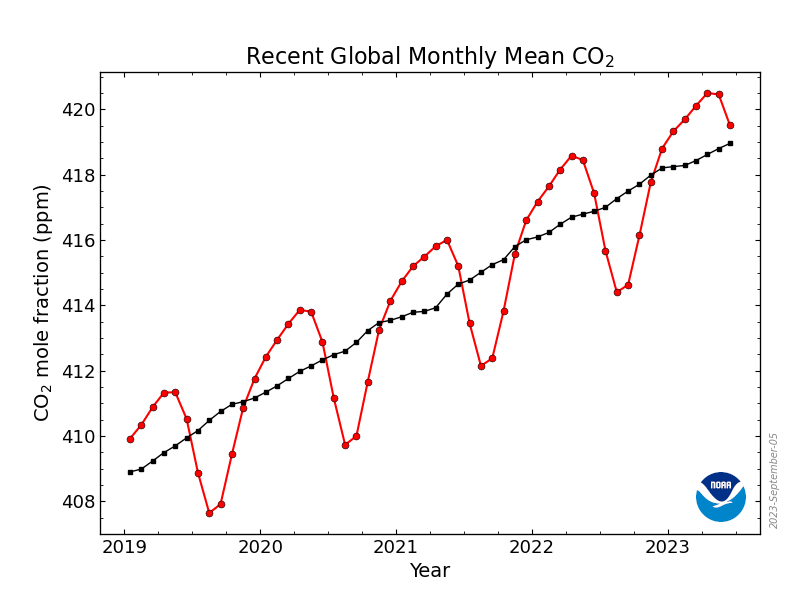
Kształt krzywej dla danych globalnych wynika głownie ze zmian sezonowych na półkulach. Na półkuli północnej jest więcej lądów, które silniej reagują na zmiany pór roku. Rośliny na lądach zmniejszają aktywność zimą i stopniowo zwiększają z początkiem wiosny, w czerwcu ich aktywność fotosyntetyczna jest już na tyle wysoka, że obniżają stężenie CO2 w powietrzu na stacjach pomiarowych na półkuli północnej, minimum przypada na jesień, kiedy to mniejsza aktywność roślin oraz rozpoczynające się procesy butwienia ponownie zwiększają poziom CO2. W ciągu roku wahania dochodzą więc do 5 ppm, natomiast z roku na rok następuje stały wzrost stężenia tego gazu o około jedną-trzecią wahań rocznych.
Selektywny odzysk uranu z morskiej wody
Uran jest pierwiastkiem dosyć rzadkim w skorupie ziemskiej, poza rudami tlenkowymi występuje w dużym rozproszeniu. Badacze z Oak Ridge University pokazali, że mimo wszystko da się go odzyskiwać nawet z takich materiałów, jak morska woda, w której występuje w średnim stężeniu około 3 miligramów na tonę.
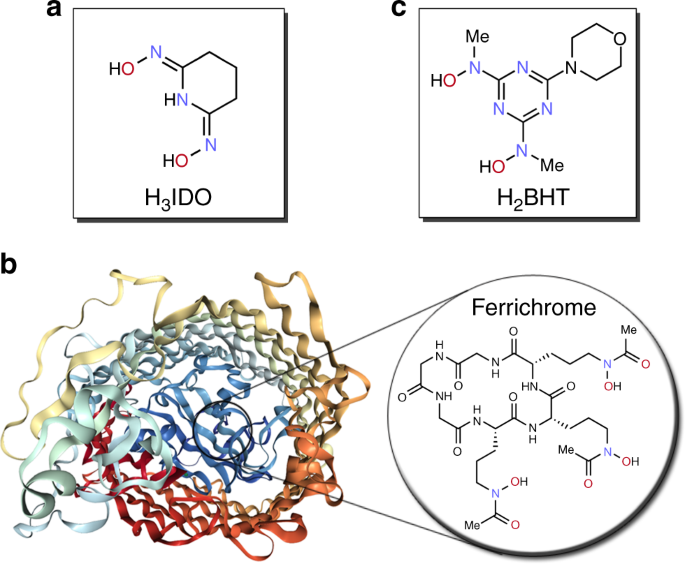
Inspiracją były bakterie i grzyby gromadzące w sobie żelazo. Posiadały białka połączone ze specjalnymi cząsteczkami, nazwanymi syderoforami, które bardzo selektywnie wyłapywały krążące w otoczeniu żelazo. Postanowiono sprawdzić, czy poprzez modyfikacje tych cząsteczek da się wytworzyć takie, które będą skutecznie wyłapywać inne pierwiastki. Metodą symulacji komputerowych i testów eksperymentalnych stworzono optymalną cząsteczkę - 2,6-bis[hydroksy(metylo)amino-4-morfolino-1,3,5-triazynę, która bardzo selektywnie pochłania jony uranowe i uranylowe. Potencjalnie więc możliwe byłoby użycie jej do odzysku tego pierwiastka. Musiałaby być osadzona na polimerowym nośniku w formie proszku, przez który można przepuścić wiele wody, aż do wysycenia cennym pierwiastkiem. [c]
--------
[a] Angel A. Martí et al. Adverse Effect of PTFE Stir Bars on the Covalent Functionalization of Carbon and Boron Nitride Nanotubes Using Billups–Birch Reduction Conditions. ACS Omega, 2019; 4 (3): 5098
[b] Rolf Güsten, Helmut Wiesemeyer, David Neufeld, Karl M. Menten, Urs U. Graf, Karl Jacobs, Bernd Klein, Oliver Ricken, Christophe Risacher & Jürgen Stutzki, Astrophysical detection of the helium hydride ion HeH+ , Nature volume 568, pages357–359 (2019)
[c] Ilja Popovs et al. Siderophore-inspired chelator hijacks uranium from aqueous medium. Nature Communications, 2019; 10 (1)







