Związek jaki został tu użyty rzeczywiście jest ciekawy, ale równocześnie bardzo prosty - w zasadzie zwykła skrobia, tylko zawinięta w małe kółko...

Omawiałem już tu kiedyś nietypowe połączenia cząsteczek "połączonych acz nie powiązanych" gdzie geometria powodowała, że dwie osobne cząsteczki tworzyły nierozerwalny układ. Teraz zajmę się przypadkiem słabszego powiązania - związku inkluzyjnego, będącego formą kompleksów typu gość-gospodarz.
W połączeniu tego rodzaju cząsteczka większa, nazywana gospodarzem, tworzy "wnękę" której kształt i rozmiar pasują do mniejszej cząsteczki "gościa". Mniejsza cząsteczka wsuwa się w większą, zagłębia we wnękę a gdy już się tam dobrze umości oddziaływania między nią a cząsteczką gospodarza tworzą kompleks, w wielu przypadkach zaskakująco trwały. Wnęka gospodarza może też nie istnieć w związku samotnym, lecz powstaje wskutek przyjmowania odpowiedniej konformacji owijającej go wokół gościa. Brzmi to bardzo intymnie.

Spośród różnych znanych układów, najbardziej popularnymi i najdłużej badanymi są cyklodekstryny. Są to fragmenty łańcucha skrobi, zamknięte w formę małych pierścieni, zawierających od 6 do ponad 30 członów glukozy połączonych wiązaniami glikozydowymi poprzez tlen.
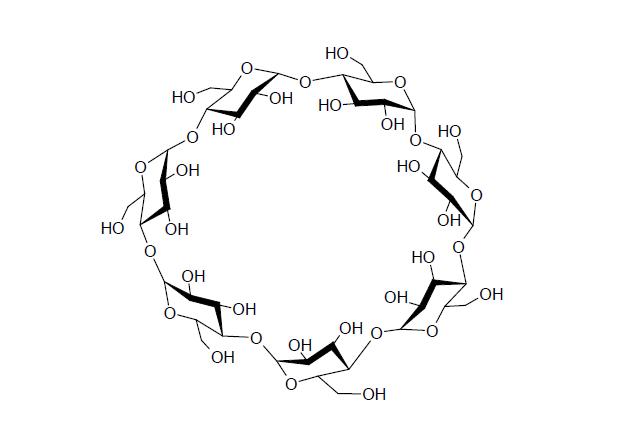
Cyklodekstryny przyjmują szczególną, nie płaską geometrię - płaszczyzny pierścieni glukozy budujących okrąg nachylają się ku sobie, przez co związek przyjmuje formę zbliżoną do ściętego stożka, lub też, jak to się często określa, do kubka z obciętym dnem. W takim układzie po stronie szerszego otworu zagęszczają się grupy hydroksylowe, przez co od tej strony cząsteczka jest hydrofilowa, natomiast po stronie otworu węższego, grupy hydroksylowe odginają się na zewnątrz, zaś okolice tego otworu i wnętrze nabierają charakteru hydrofobowego:

Alfa cyklodekstryna tworzy "kubek" o wysokości 0,78 nanometrów i średnicy wewnętrznej 0,57 nm; beta cyklodekstryna przy tej samej wysokości ma wnękę o średnicy 0,78 nm a gamma 0,95. Wielkości tych wnęk determinują rodzaj cząsteczek które mogą do nich wniknąć - zbyt duże się nie zmieszczą, zaś bardzo małe będą słabiej oddziaływały.
Jeśli chodzi o rodzaj sił wciągających cząsteczki do wnętrza cyklodekstryny, to oprócz sił van deer Walsa znaczenie ma tu też adsorbcja hydrofobowa. Cząsteczka hydrofobowa słabo oddziałuje z wodą i podobnymi do niej rozpuszczalnikami, efekty oddziaływań między cząsteczkami wody prowadzą do odpychania grupy hydrofobowej. W tej sytuacji cząsteczki hydrofobowe będą dążyły do utworzenia agregatów, zaś w naszym przypadku mała cząsteczka hydrofobowa będzie wpychana do mającego takie właściwości wnętrza cyklodekstryny.
Cząsteczki zawierające fragmenty z elektroujemnymi niemetalami mogą dodatkowo tworzyć wiązania wodorowe z grupami -OH na obrzeżu. Ponadto możliwe jest tworzenie kompleksów koordynacyjnych. Cyklodekstryny to jedne z nielicznych cząsteczek organicznych kompleksujących aniony. Gdy hydrofobowa cząsteczka jest dłuższa niż wynosi głębokość pierścienia, możliwe jest dołączenie drugiego. Tak powstały kompleks o stosunku 1:2 nazywa się molekularną kapsułką lub też niezupełnie poprawnie, mikrokapsułką.

Wykazano powstawanie kompleksów z bardzo dużą ilością cząsteczek organicznych i nieorganicznych, nieraz całkiem sporych, na przykład fullereny, i szybko zaczęto ten fakt wykorzystywać. Zamknięte w molekularnej kapsułce związki przechodzą do roztworu w wodzie oraz są w pewnym stopniu chronione przed zewnętrznymi wpływami, stąd chętne użycie cyklodekstryn jako nośnika substancji zapachowych i smakowych dodawanych do żywności. Sama cyklodekstryna ma na liście dodatków oznaczenie E459. Może być też używana do stabilizacji składników odżywczych i witamin, chroniąc je przed utlenieniem w żywności suchej. Udało się w ten sposób stworzyć rozpuszczalną formę kurkuminy, która w normalnych warunkach jest słabo rozpuszczalna.
Zastosowania medyczne
Jednym z ciekawszych przypadków takiego kompleksowania, który znalazł zastosowanie w medycynie, jest tworzenie połączeń z cholesterolem. Cząsteczka cholesterolu jest generalnie hydrofobowa i słabo rozpuszczalna w wodzie natomiast dobrze w tłuszczach. Jej rozmiar i kształt idealnie pasuje do alfa-cyklodekstryny. Po dodaniu cyklodekstryn do żywności duża część cholesterolu zostaje związana co utrudnia jego wchłanianie. W taki sposób produkuje się jedzenie niskocholesterolowe.
Obecnie testuje się pochodne cyklodekstryn jako lek na chorobę Niemanna-Picka typu C. Choroba ta, wywołana pewnymi mutacjami, powoduje zaburzenia w transporcie substancji do komórek, wywołując gromadzenie się cholesterolu w lizosomach i sfingolipidów w neuronach. Prowadzi to do zaburzeń czynności wątroby i trzustki, oraz objawów neurologicznych, w przypadku dzieci wywołujących niepełnosprawność i opóźnienie umysłowe, a w przypadku osób starszych szybko postępującą demencję, głuchotę, zaburzenia psychiczne, padaczki. Podobieństwo objawów powoduje, że czasem nazywa się ją "dziecięcym Alzheimerem".
W 2009 roku zezwolono na eksperymentalne użycie hydroksypropylowej pochodnej beta-cyklodekstryny do łagodzenia przebiegu choroby u sióstr bliźniaczek[1], gdyż usuwa cholesterol z lizosomów, co powinno ograniczyć postęp choroby. Potem zastosowano ją jeszcze u kilkunastu pacjentów ale nie ma jeszcze ostatecznych wniosków na ile jest to sposób skuteczny. Pewne niedawne badanie na kilku pacjentach sugeruje spowolnienie rozwoju choroby. [2] Substancja jest w takich zastosowaniu podawana w formie roztworu do płynu mózgowo-rdzeniowego.

Inny przykład to Sugammadeks, lek odwracający blokadę mięśniowo-nerwową u osób którym podano leki zwiotczające na przykład przy znieczuleniu ogólnym. Jest to cząsteczka gamma-cyklodekstryny zmodyfikowana przez dodanie grup sulfanylopropionowych. Rodzaj grup i ich długość dobrano tak, aby cząsteczka idealnie pasowała do środka zwiotczającego rokuronium. Początkowo miał to być nośnik leku ułatwiający rozpuszczanie w wodzie, ale po stwierdzeniu wyjątkowo dużej siły kompleksowania, zaczęto stosować zmodyfikowaną cyklodekstrynę do usuwania środka z ustroju. Po wstrzyknięciu do krwioobiegu, sugammadeks kompleksuje rokuronium, w związku z tym związek ten zaczyna być oddawany przez tkanki co znosi działanie zwiotczające.
A co z alkoholem?
Etanol jest małą cząsteczką organiczną z jednym końcem o pewnych właściwościach lipofilowych, i już dawno stwierdzono, że w odpowiednich warunkach możliwe jest stworzenie połączenia inkluzyjnego z cyklodeksytryną, które jednak rozpadało się pod wpływem wody. Pierwsze próby zastosowań spożywczych miały miejsce w latach 70. ale najwyraźniej nie były zbyt udane, dlatego dla przeciętnego konsumenta wynalazek zaistniał dopiero w ostatnich latach. Najczęściej spotykane użycie, to napoje typu "grzaniec" - te dostępne na polskim rynku zawierają enkapsulowany alkohol w ilości odpowiadającej stężeniu 0,5% w gotowym napoju (w zasadzie więc są to ilości dla aromatu).
W Europie dostępne są napoje w proszku o smaku szampana czy wina z dodatkiem alkoholu w ilości wystarczającej, aby się upić.
Proszek taki składa się z drobnych cząstek zawierających wewnątrz masę kompleksu cyklodekstryna-alkohol, otoczoną warstewką ochronną liniowych dekstryn, chroniących wnętrze przed parowaniem. Pył może zawierać do 30% alkoholu.
Cyklodekstryny spożyte doustnie nie wywołują szkodliwych skutków, są częściowo trawione tak samo jak zwykła skrobia. Ze względu na rozmiar cząsteczek nie są wchłaniane do organizmu. Testuje się je jako środek obniżający poziom cholesterolu, zażywany w dawkach kilkugramowych.[3]
---------
* https://en.wikipedia.org/wiki/Alcohol_powder
* https://en.wikipedia.org/wiki/Cyclodextrin
[1] http://addiandcassi.com/walgreens-support-twins-niemann-pick-type-receive-cyclodextrin-treatments-home/
[2] https://www.sciencedaily.com/releases/2017/08/170810192740.htm
[3] https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4941029/








































{kind=link}