
Historia
Szkocki chirurg dr Henry Faulds był ciekawą postacią. W ramach misji prezbiterianów w latach 70. XIX wieku udał się do Japonii gdzie wywarł duży wpływ na rozwój nowoczesnej chirurgii w tym kraju. Zakładał szpitale w których stosował antyseptyczny reżim Listera, zapobiegający zakażeniom. Założył pierwszy na wyspach zakład opiekujący się niewidomymi, propagował higienę i przyczynił się do zakończenia kilku epidemii. Napisał dwie książki podróżnicze a jego szpital w Tokio był uważany za najlepsza azjatycką placówkę zdrowotną.
W trakcie tych wszystkich zajęć miał jednak czas aby zajmować się różnymi drobnostkami. Na przykład pomagał w wykopaliskach przyjacielowi, Edwardowi Morse'owi, który przekopując starożytne kopce i ruiny segregował odnalezioną ceramikę, starając się określić osobne okresy kulturowe. Gdy przeglądali szczątki rozbitych waz, talerzy, dzbanów i drobnych przedmiotów, zwrócili też uwagę na odciśnięte ślady palców starożytnych rzemieślników, którzy kształtowali miękki materiał a po których po tysiącach lat jako jedyny ślad pozostał odcisk delikatnych linii. Faulds zaczął się wówczas przyglądać własnym palcom, i porównywał ich wygląd z palcami innych ludzi. Stwierdził, że w szczegółach różnią się one od siebie na tyle dobrze, że po samych śladach dałoby się, jak sądził, stwierdzić kto je zostawił. I być może uwaga ta skończyłaby się najwyżej drobnym artykułem czy też jakąś wzmianką w kolejnej książce o swej praktyce lekarskiej, gdyby nie sprawa która wręcz zmusiła go do działania.
Oto doktor stwierdził, że ktoś podkrada mu alkohol z szafki. Sprawca pozostawił na butelce wyraźne, tłuste odciski. Ponieważ do szafki miało dostęp tylko kilka osób, poprosił je aby pozostawiły mu odbitki opuszków swych palców umoczonych w atramencie. Po wskazaniu, że jego odciski wyglądają tam samo jak te z butelki, jeden z studentów przyznał się. Nieco później jednego z lekarzy oskarżono o kradzież z włamaniem do domu, gdzie sprawca, wspinając się po okopconym sznurze, pozostawił na ścianie odcisk całej dłoni. Wprawdzie nie było jeszcze wówczas pewne czy wzory się nie powtarzają, ale wszyscy się zgodzili, że skóra na dłoniach lekarza nie mogła zmienić się w ciągu jednego dnia, gdy więc Faulds pokazał że odcisk nie pasuje do dłoni oskarżonego, policja uznała siłę dowodu i uwolniła go.
Zachęcony tym sukcesem Faulds, napisał artykuł opisujący swe odkrycia,. zawierający wyraźne wskazanie, że dysponując obszerną bazą takich śladów można by ułatwić rozwiązywanie zagadek kryminalnych. Gdy zaś w roku 1880 opublikował go Nature, okazało się ze Faulds nie był pierwszy. Już w latach 60. XIX wieku brytyjski urzędnik sir William Hershel, urzędujący w kolonii w Indiach, uznał że najlepszym sposobem osłabienia fali fałszerstw, będzie nakazywanie wypełniającym dokumenty aby "podpisywali" je przystawiając obok tekstu odcisk całej dłoni. Ułatwiało to prace w przypadku niepiśmiennych, którzy nie mieli wyrobionego podpisu, zwiększało strach oszustów których identyfikacja byłaby szybsza i odwoływało się do przesądnego powiązania śladów z osobą.

Po pewnym czasie, przekonawszy się że na żadnym z tysięcy odcisków wzory się nie powtarzają, poprzestał na odbitkach palca środkowego i kciuka.
Po artykule w Nature, Hershel zgłosił się jako wcześniejszy odkrywca tej metody identyfikacji, zaś pomiędzy obydwoma panami rozpoczął się zacięty spór o pierwszeństwo, trwający aż do XX wieku. Faulds próbował zainteresować swymi wynikami Scotland Yard, ale rewelacje te przyjęto wówczas chłodno, przedkładając nad odciski będący nowością system antropometryczny Bertillona, opisujący przestępców a pomocą danych wielkości, długości i rozstawy charakterystycznych cech budowy fizycznej
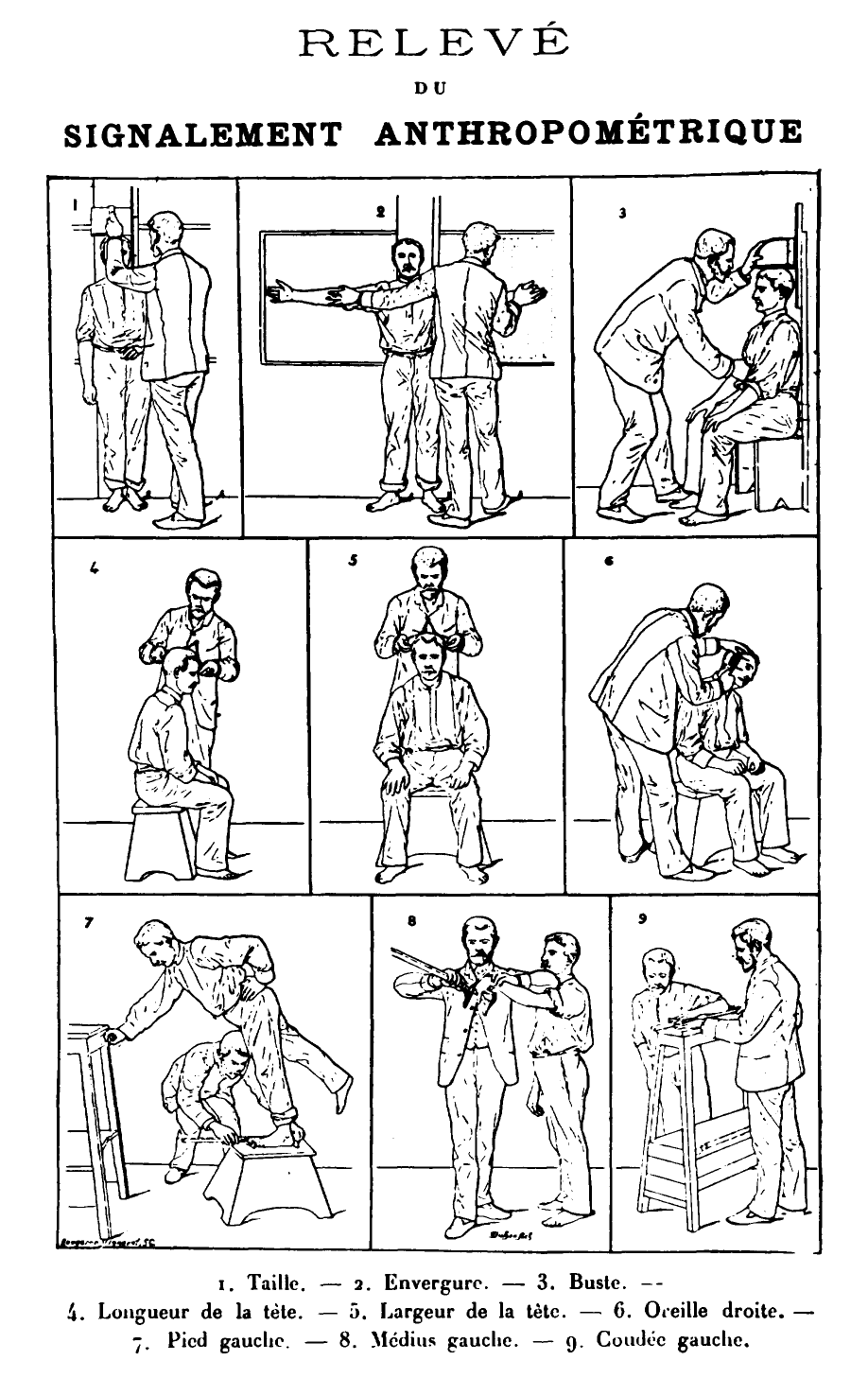
Jeszcze przed publikacją Faulds opisał swe badania w liście do Karola Darwina, którego jednak niespecjalnie one zainteresowały. Przekazał jednak list swemu kuzynowi Francisowi Galtonowi, który zajął się tą sprawą sądząc, że będzie w stanie znaleźć jakieś specyficzne cechy rasowe i dziedziczne, mogące pozwalać ocenić cechy charakteru, umysłowości czy wyglądu. Nie udało mu się to, ale uczynił co innego - opierając się na kartach Hershela i własnych badaniach wykazał unikalność linii papilarnych, a tym samym przydatność w identyfikacji. On też opisał występowanie we wzorach charakterystycznych punktów - minucji - będących miejscami zakończeń, skrzyżowań lub rozwidleń linii. Pozwoliło to lepiej porównywać ze sobą odciski. Owocem badań była książka wydana w 1892, a także liczne artykuły.
Przydatność nowej metody ujawniła się bardzo szybko, bo już w tym samym roku. 19 czerwca 1892 roku w argentyńskiej miejscowości Necochea popełnione zostaje brutalne morderstwo - nieznany sprawca zabija nożem dwójkę małych dzieci 27-letniej Franceski Rojas. Oskarża ona o zabójstwo swego sąsiada, którego zaloty odrzucała od dłuższego czasu. Tamtejsza policja bierze go na przesłuchanie, bardzo długie i brutalne, ale nie dochodzi do rozstrzygnięcia - podejrzany nie przyznaje się zaś jego znajomi potwierdzają alibi. Równocześnie wyjawia, że matka zabitych dzieci znalazła ostatnio narzeczonego, który, jak podsłuchał z jej narzekań, nie chce się z nią ożenić "dopuki ma te przeklęte bachory". Poszlak mogących potwierdzić którąś z teorii brakowało.Śledczy znaleźli się w impasie. Inspektor prowadzący sprawę odkrywa jednak, że mimo upływu kilku dni na futrynie drzwi zachował się bardzo wyraźny krwawy ślad palca sprawcy, i przypomina sobie Juliana Vuceticha, który mówił mu niedawno o tego typu dowodach.
Vucetich, będący urzędnikiem policyjnym z centrali, był nieoczekiwanie Chorwatem, a przy tym człowiekiem bardzo postępowym. Już w poprzednim roku natknął się na artykuły Galtona, po których ściągnął publikację Fauldsa i zaciekawiony nakazał policjantom pobierać odbitki palców od zatrzymywanych więźniów, mając nadzieję zestawienia bazy pomocnej w razie ich recydywy. Jego inspektor badający sprawę w Necochea pamiętając o tym, odpiłował od futryny kawałek ze śladem, po czym kazał zostawić odbitki palców wszystkim zamieszanym w sprawę. Wprawdzie techniki analizy były wówczas bardzo prymitywne, ale nie trzeba było wielkiego rozeznania, aby zobaczyć, że ślad pasuje do linii papilarnych matki.
Po okazaniu dowodów, Francesca Rojas przyznała się. Została skazana na dożywocie. Był to pierwszy taki przypadek w historii kryminalistyki.

Jak powstają ślady linii papilarnych?
Skóra jest stale zwilżana potem wydzielanym przez odpowiednie gruczoły. Pot zawiera głównie wodę, sole mineralne i proste związki organiczne, w tym węglowodany, aminokwasy i kwasy tłuszczowe. W dodatku skóra jest natłuszczana łojem. Mieszanka obu substancji tworzy na powierzchni opuszek palców cienką warstewkę. Gdy dotykamy jakiejś powierzchni, substancja potowo-tłuszczowa zostaje na nią naniesiona, ale z oczywistych względów tylko ta pokrywająca wystające listewki linii papilarnych nie zaś ta wewnątrz głębokich rowków między nimi. Na powierzchni pozostaje więc ślad listewek skórnych.
Jeszcze kwestia terminologii - przyjęło się powszechnie mówić o odciskach palców, jednak specjaliści uznają ten termin za niedokładny. Odcisk powstaje w wyniku odciskania kształtu w miękkim materiale, toteż za odcisk palca możemy uznać ślad pozostawiony na przykład w glinie, plastelinie, wosku czy nawet grubej warstwie brudu - i takie są też znane kryminalistyce. Inaczej wygląda rzecz gdy chodzi o dotykanie powierzchni twardych, wówczas pozostaje jedynie odbicie wzoru linii, ale nie wgłąb materiału. Dlatego za poprawny uważa się termin odbitki palców czy odbitki linii papilarnych. Można też mówić o śladach daktyloskopijnych czy wreszcie śladach palców.
Powstawanie odbitki linii papilarnych jest podobne do odbijania druku ze wzoru czcionki czy drzeworytu, co znalazło odzwierciedlenie w angielskim określeniu "fingerprint" czyli dosłownie "palcodruk".
Linie na opuszku palców są na dłuższą metę nieusuwalne - wprawdzie niektórzy przestępcy próbowali takich metod jak wytrawianie naskórka kwasem czy ścieranie, ale wraz z odradzaniem skóry, linie powracały. Potrafią odtworzyć się nawet po odcięciu skóry, zwłaszcza że końcówki palców mają dużą skłonność do odrastania. Kiedyś przy wycinaniu chwastów na działce zdarzył mi się przykry wypadek w wyniku którego odciąłem sobie nożem pół opuszka palca serdecznego. Miejsce to zagoiło się tak że dziś nie da się zauważyć śladów po cięciu. Z drugiej strony na jednym z kciuków w odcisku daje się zauważyć cieniutką bliznę - ślad po głębokim skaleczeniu z wczesnego dzieciństwa.
W sytuacji gdy ślad jest odciśnięty w miękkim materiale lub odwzorowany substancją o wyraźnym kolorze, a więc krwią czy smarem, nie ma problemu w jego znalezieniu i skopiowaniu. Wiele jednak odbitek jest niewidocznych, zwłaszcza na matowej powierzchni. Standardowym sposobem ujawniania tych śladów utajonych, jest nanoszenie drobnego proszku mającego większą skłonność do przylepiania się do potowo-tłuszczowej substancji niż do podłoża, zazwyczaj przy pomocy pędzelka o delikatnym włosiu. Standardowo używanymi proszkami są różne odmiany sadze, proszki metaliczne czy tlenki metali. Ciekawym przypadkiem są proszki magnetyczne, nakładane na powierzchnię w formie "pęczka" przyklejonego do magnesu, co zapewnia bardziej delikatne naniesienie bez ryzyka zostawienia rys po włoskach

Jednak poza tymi prostymi technikami, stosowane mogą być specyficzne odczynniki ujawniające utajnione ślady, zwłaszcza te starsze, które po wyschnięciu straciły lepkość i słabo przyklejają do siebie proszek. Jakie są to odczynniki? Pisałem już kiedyś o chemicznych testach na wykrycie krwi, teraz więc skupię się na chemicznych odczynnikach pozwalających na ujawnienie i utrwalenie niewidocznych odbitek daktyloskopowych nawet po upływie wielu lat
Jod
Użycie par jodu do ujawnienia niewidocznych odcisków było pierwszą nie proszkową metodą, znaną już od 1863 roku ale w kryminalistyce użytą dopiero na początku XX wieku. Zasada działania opiera się na niezwykle prostym mechanizmie - stały jod, mający postać grafitowego proszku, powoli paruje, zwłaszcza po lekkim podgrzaniu. Jego opary chętniej rozpuszczają się i gromadzą w tłuszczowym śladzie odcisku palca niż na większości badanych powierzchni. W efekcie po pewnym czasie odcisk zabarwia się na żółto lub pomarańczowo:

Ninhydryna
Ninhydryna formalnie rzecz biorąc może być uznana za wodzian, czyli pochodną ketonu, o dużej reaktywności za sprawą oddziaływań dwóch sąsiadujących takich grup. Chętnie w związku z tym reaguje z aminami a także aminokwasami, będącymi składnikiem śladu tłuszczowego odcisku palca, tworząc w szeregu reakcji produkty o purpurowej barwie:
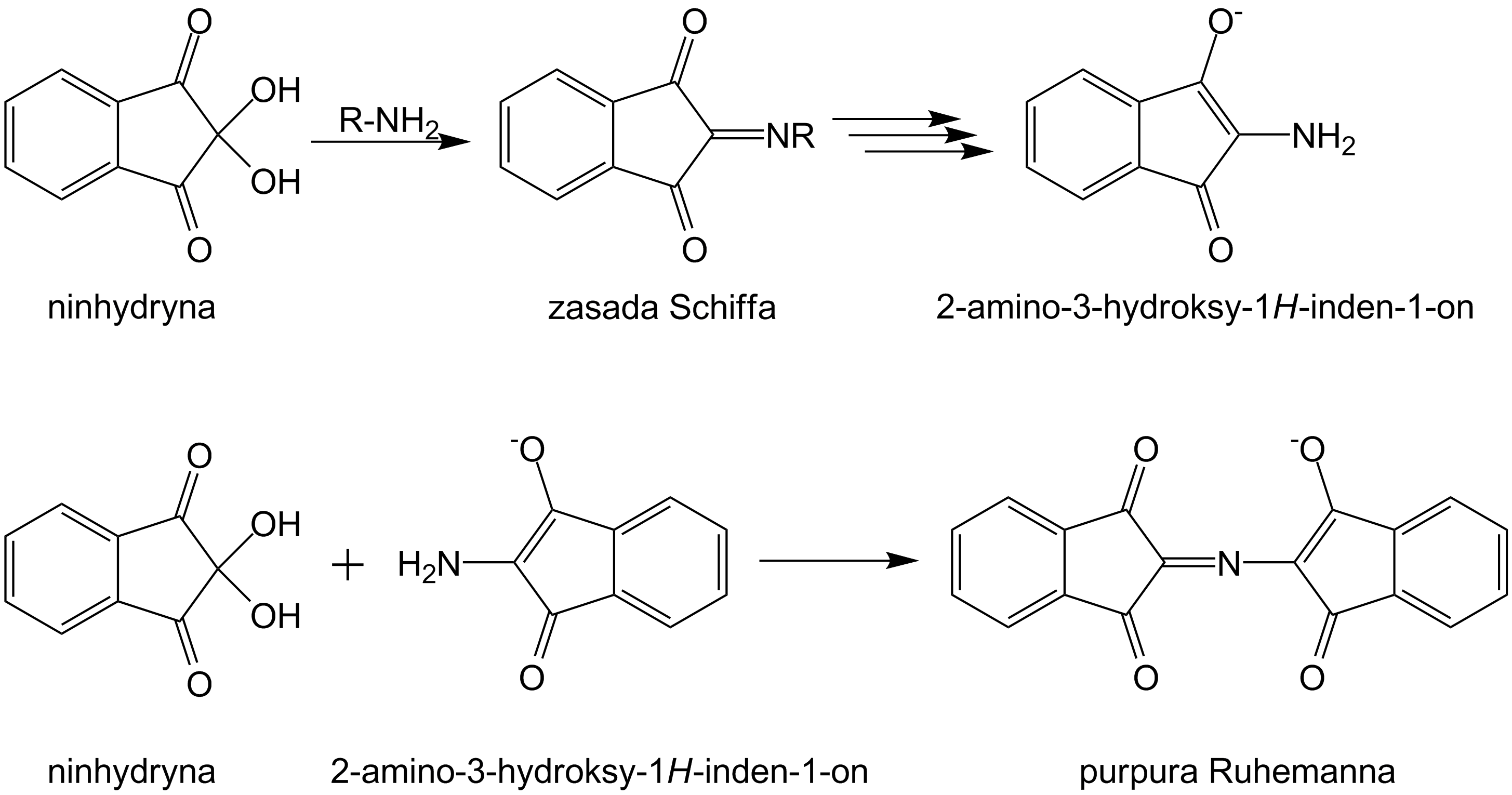
Tylko z niektórymi aminokwasami daje inne zabarwienie, a z aminami drugorzędowymi pomarańczowe sole. Znana w chemii do oznaczania aminokwasów i białek, w kryminalistyce znalazła zastosowanie w latach 50. do ujawniania odcisków na materiałach porowatych i chłonnych, jak papier czy drewno, na których wchodzący w drobne szczelinki proszek zupełnie zaciemniałby obraz.
Ninhydryna rozpuszczona w bezwodnym alkoholu musi być napryskana na badaną powierzchnię przy pomocy spryskiwacza dającego drobne kropelki lub przy pomocy spreju bo i takie zestawy stworzono. Ma bardzo drażniący zapach i prowokuje kaszel dlatego lepiej robić to przy dobrej wentylacji. Powierzchnia powinna być równo pokryta ale nie zmoczona. Aby zaszła reakcja pokrytą powierzchnię należy ogrzać - gdy tą metodą ujawniałem chromatogram bibułowy aminokwasów, wystarczało potraktowane suszarką nastawioną na grzanie, choć dla papieru dobrym sposobem mogłoby być użycie żelazka.

Z oczywistych względów metoda nie nadaje się do powierzchni ze skóry naturalnej, która zabarwiłaby się nam cała. W przypadku papieru nakredowanego, o lekko zasadowym odczynie, reakcja może bądź nie zajść bądź dać słabe zabarwienie. Zwykle przeciwdziała się temu przez dodatek kwasu octowego do roztworu.
Częstą metodą obróbki ujawnionych odbitek jest potraktowanie ich eterowo-alkoholowym roztworem soli cynku. Tworzy on z purpurowym związkiem kompleks o kolorze słabo pomarańczowym, ale za to świecący w ultrafiolecie, co może mieć znaczenie w przypadku gdy powierzchnia badana jest kolorowa. W późniejszych latach wymyślono szereg pochodnych ninhydryny oraz związków o podobnej reaktywności, mających zastosowanie w szczególnych przypadkach. Jednym z nich jest Diazafluorenon (DFO).
Związek ten został zastosowany w kryminalistyce stosunkowo niedawno. Pod względem budowy oraz działania jest podobny do ninhydryny dając przy tym dość słabe, pomarańczowe zabarwienie, ma jednak pewną cenną właściwość - w świetle niebieskim fluoryzuje na żółto, co pozwala zauważyć ujawnione ślady nawet gdy są one bardzo słabe, działa więc podobnie do cynkowego kompleksu ninhydryny ale jest prostszy w użyciu.
Dobre cechy ninhydryny i DFO łączy w sobie inna pochodna, 5-metylotioninhydryna(5-MTN). Ze śladami aminokwasów daje purpurowe zabarwnienia, lecz po potraktowaniu solami cynku staje się ono tylko ciemniejsze. Pod wpływem zielonego światła cynkowy kompleks fluoryzuje na żółto co obserwuje się przez filtr czerwony. Istnieje jeszcze specjalna wersja ninhydryny do zastosowania na papierze termicznym (paragony sklepowe).[2] [3]
Super klej
Często dziś używaną metodą ujawniania niewidocznych odcisków na twardych, gładkich powierzchniach jest metoda cyjanoakrylowa. Odkryto ją w dużej mierze przypadkowo - japoński technik kryminalistyki Fuseo Matsumur zajmował się śladami mikrowłókien i włosów, które zbierał i zabezpieczał na szklanych płytkach pokrytych warstewką szybko schnącego kleju. Płytki przechowywał potem w pojemniku z przegródkami, tak aby płytki nie stykały się ze sobą i aby nie zanieczyszczały ich włókna spoza miejsca zbrodni. Przeglądając płytki zauważył w 1977 roku, że na odwrotnej stronie po pewnym czasie przechowywania ujawniają się odciski jego palców, dając niezmywalne białe ślady. Zainteresował tym kolegów i wkrótce Japończycy opracowali metodę w takiej formie jaką znamy dziś.
Wszystkie szybko schnące superkleje cyjanoakrylowe, zawierają głównie takie związki jak 2-cyjanoakrylan metylu, etylu lub butylu. Są to zatem estry pochodnej kwasu akrylowego podstawionej grupą nitrylową przy drugim węglu. Taka struktura jest bardzo nietrwała, grupa nitrylowa z jednej strony a estrowa z drugiej, odciągają elektrony z fragmentu łańcucha z wiązaniem podwójnym. W efekcie ten fragment staje się podatny na atak grup nukleofilowych, a więc aktywnych cząstek z parą elektronową. Przykładem takiego nukleofila może być grupa hydroksylowa powstająca z dysocjacji wody. Jej przyłączenie do końcówki wiązania podwójnego powoduje jego pęknięcie i wytworzenie karboanionu. Ten jest dobrym nukleofilem i reaguje z kolejną cząsteczką akrylanu. Zapoczątkowana śladami jonów hydroksylowych reakcja biegnie dalej sama, tworząc polimer dobrze wiążący ze sobą powierzchnie klejone.

A co to ma wspólnego z ujawnianiem odcisków palców?
Monomery cyjanoakrylowe są lotne, co zresztą jest jedną z ich wad, bowiem w większych stężeniach stają się trujące. Monomer akrylowy nie powinien reagować z większościami powierzchni, może natomiast wchodzić w reakcję z aminokwasami, lipidami i cukrami substancji śladów palców, zwłaszcza w obecności wilgoci.


Ujawnione tą metodą ślady są białe lub kremowe, aby zwiększyć kontrast i lepiej je uwidocznić, na przykład proszkami; często stosuje się fluorescencyjne barwniki chętnie łączące się z substancją polimerowo-tłuszczową, na przykład Basic Yellow 40 świecący w ultrafiolecie na żółto-zielono, Safranina O świecąca w zielonym świetle na czerwono albo Rodamina 6G świecąca na żółto. Używa się kilkunastu takich preparatów, zależnie od rodzaju powierzchni i dostępności[4][5]
Azotan srebra
Ta metoda nie jest zbyt często używana, choć znano ją już w XIX wieku. Właściwie używa się jej do poszukiwania śladów tak starych że pozostałe metody nie są w stanie ich ujawnić, ale dopiero na końcu, jest bowiem niszcząca. Jako pierwsza ze śladu ulatnia się woda, po niej krótkołańcuchowe kwasy tłuszczone (skóra dziecka wytwarza tylko takie kwasy przez co odciski palców dziecka dość szybko zanikają) na koniec rozkładowi ulegają aminokwasy. Co więc może pozostać na powierzchni, gdy wszystko inne już zniknie? Sole mineralne a przede wszystkim chlorek sodu obecny w pocie.
Badane powierzchnie należy spryskać azotanem srebra ale tak aby nie były zupełnie zmoczone. Jony srebra w reakcji z jonami chlorkowymi dadzą biały, nierozpuszczalny osad chlorku srebra, który po wystawieniu na słońce ciemnieje pozostawiając ciemne plamy. Takie same plamy powstaną na ubraniu oraz naszej skórze, dlatego przy użyciu tego związku można się na prawdę mocno i dosyć trwale pobrudzić. Metoda nadaje się do papieru i skóry, nie sprawdza się przy drewnie i tkaninach. Jeśli badana powierzchnia była wystawiona na działanie wody, ślady zostaną zmyte. Z oczywistych względów przeszkadzają tu sole mineralne których obecność zaciemnia tło. [6]
Gencjana
Fiolet krystaliczny lub metylowy, to mieszanina związków o budowie podobnej do fenoloftaleiny. Dawniej używana do farbowania wełny czy jako barwnik ołówka kopiowego, dziś też niekiedy do odkażania ran. Jest barwnikiem lipofilowym, w związku z tym ma skłonność do wchłaniania przez tłuszcze. Na tym też opiera się jej działanie.
W kryminalistyce znalazła zastosowanie do uwidaczniania odcisków na materiałach lepkich, jak na przykład taśmy klejące, etykietki itp. Badany przedmiot zanurza się w jej ok. 2% roztworze na dwie minuty, po czym spłukuje czystą wodą. Odbitki linii papilarnych zabarwiają się wówczas na fioletowo[7]
Czerń Sudan
Ciemny barwnik o właściwościach lipofilowych, działający tak samo jak gencjana - chętniej absorbuje się w tłuszczowym śladzie niż w podłożu. Ma zastosowanie na materiałach klejących, zatłuszczonych lub woskowatych, na przykład nawoskowany papier czy folie spożywcze. Może być też użyta do zabarwienia śladów ujawnionych metodą cyjanoakrylową na przykład w przypadkach gdy dotyczą one powierzchni jasnych, nie chłonnych i fluoryzujących.[8]
Lumicyano?
Najnowsza technika kryminalistyczna, jest połączeniem kilku wartościowych cech. Tak jak pochodne ninhydryny miały łączyć zdolność ujawniania kontrastowych śladów z fluorescencją, tak lumicyano łączy technikę cyjanoakrylową z luminescencyjną bez potrzeby stosowania dodatkowych odczynników. Pomysł jest prosty - do cząsteczki cyjanoakrylanu podczepiono odpowiednią grupę, w tym przypadku jest to tetrazyna - pięciokątny pierścień z czterema atomami azotu.
Tak samo jak superklej, po ogrzaniu paruje i polimeryzuje w śladzie tłuszczowym. Ujawniony kremowy odcisk fluoryzuje w ultrafiolecie.[9]
Technika została już przetestowana w laboratoriach kryminalistycznych. Wyniki badań pojawiły się w tym roku.
-------
* http://en.wikipedia.org/wiki/Fingerprint
* http://onin.com/fp/fphistory.html
* http://www.bvda.com/EN/sect1/en_1_6a.html
* http://en.wikipedia.org/wiki/Henry_Faulds
* http://en.wikipedia.org/wiki/Sir_William_Herschel,_2nd_Baronet
* http://en.wikipedia.org/wiki/Francis_Galton
* http://en.wikipedia.org/wiki/Juan_Vucetich
* http://en.wikipedia.org/wiki/Francisca_Rojas
[1] http://makezine.com/forensics-laboratory-82-revealing-l/
[2] www.viewsfromscience.com/documents/webpages/led_fluorescence_p7.html
[3] http://makezine.com/forensics-laboratory-83-revealing-l/
[4] http://www.bvda.com/EN/sect1/en_1_9a.html
[5] http://makezine.com/projects/fingerprinting-with-super-glue/
[6] http://makezine.com/laboratory-84-revealing-latent-fing/
[7] http://makezine.com/laboratory-86-revealing-latent-fing/
[8] http://www.bvda.com/EN/sect1/en_1_12a.html










