był gwałtownikiem i idealnie nadawałby się na bohatera historycznej tragedii. Pochodził ze znanej i szanowanej belgijskiej rodziny szlacheckiej, mającej majątki w zamorskich koloniach. Wedle rodzinnej opowieści urodził się na statku miotanym sztormem podczas podróży do Malezji, co nadało mu gwałtowny charakter ze skłonnością do awantur. Wychowywany przez malezyjskie matki dostał kiedyś do zjedzenia cząstkę lwiego serca, aby nabrał odwagi. Gdy jego ojciec stracił lukratywne stanowisko w kolonii na Jawie, przeniósł się wraz z nim do Arkansas, gdzie odpierając ataki Indian pomagał w zakładaniu kolejnej kolonii. Włóczył się z traperami i polował na dziką zwierzynę, jednak długo nie wytrzymał na prowincji. Gdy wreszcie w latach 40. XIX wieku wylądował w Belgii był bogaty w doświadczenia, lecz ubogi majątkowo.
Dzięki powiązaniom rodzinnym był jednak wystarczająco "na poziomie" aby móc ożenić się z
Lidią Fougnies du Bois, z bogatej rodziny kupieckiej, która dorobiła się na handlu towarami kolonijnymi.
Jak mówiono potem, Lidia miała romantyczne usposobienie, nie dosyć, że zaczytywała się w młodości opowieściami o wielkich miłościach, ale wreszcie sama zaczęła pisać romanse, nie osiągając jednak większego sukcesu.
Żona wniosła pokaźny posag, który pomógł w utrzymaniu czwórki dzieci i średniowiecznego zamku Bitremont, jednak pieniądze szybko się kończyły. Hrabiostwo musieli zwolnić część służby i ograniczyć niektóre rozrywki aż wreszcie wyprzedawać ziemię. Pewną nadzieję na poprawę sytuacji dawał spadek po ojcu Lidii, właścicielu potężnego majątku. Niestety gdy otworzono testament okazało się, że większość swego majątku przekazał synowi Gustawowi. Był to człowiek chorowity, od dłuższego czasu narzekający na zdrowie, z powodu amputowanej stopy mający problemy z poruszaniem się, toteż małżonkowie mieli nadzieję na jego szybką śmierć i przekazanie spadku.
Jednak w 1850 roku Gustaw ogłosił, że się zaręcza.
Łatwo domyśleć się reakcji de Bocarme'ów - jeśli właściciel większości majątku ożeni się, w razie jego śmierci pieniądze przypadną żonie. Nie była to zbyt miła perspektywa.
20 listopada Gustaw przybywa do Bitremontu na zaproszenie siostry. Lidia musi niestety odmówić jego prośbie uczestnictwa w ślubie, hrabiostwo będą bowiem w tym czasie wyjeżdżać. Trudno, zdarza się. Wizyta przebiegała zupełnie spokojnie aż do kolacji, kiedy to hrabia wyprosił służbę i dzieci, chcąc zjeść ją tylko z siostrą i szwagrem. Drzwi zostają zamknięte. Atmosfera tej chwili jest najzupełniej odpowiednia, noc była to bowiem burzliwa i deszczowa.
Służący mimo nakazu oddalenia się, kręci się na korytarzu pod jadalnią. Dzięki temu wyraźnie słyszy rumor wewnątrz, trzask przewracanego krzesła i okrzyk "Przebacz!". Po kilku minutach drzwi otwierają się, zaś Hipolit w zmiętym ubraniu oznajmia zdumionej służbie, że szwagier Gustaw nagle zmarł.
Miał podobno nagle zacząć skarżyć się na ból głowy. Potem osunął się z krzesła a jego ciałem wstrząsały konwulsje. Następnie zwiotczał i przestał oddychać. To z pewnością nagły udar mózgu - stwierdził hrabia, po czym poprosił służących o wiadro octu aby móc obmyć zwłoki i przygotować do pogrzebu. Równocześnie bardzo stanowczo zakazał komukolwiek ze służby udawać się do miasta. Gdy hrabia i hrabina obmywali octem trupa, a następnie skrobali zaplamione deski podłogi, cały czas zapewniając że to tylko tragiczny wypadek, służący opowiedział o wszystkim mieszkającemu obok księdzu. Ten, nie czując się związany żądaniami hrabiego, udał się co prędzej do miasta. Następnego dnia na zamek przybył sędzia aby rzecz zbadać, albowiem sami przyznacie, że okoliczności tej śmierci nie mogły być już chyba bardziej podejrzane.
Zarówno na dłoniach hrabiego jak i na ubraniu zmarłego widać było ślady szamotaniny a nawet głębokie ugryzienie - to skutek konwulsji, tłumaczył Hipolit. Na twarzy i języku zmarłego widać było oparzenia chemiczne - to przez wymioty, tłumaczył hrabia. Sekcja zwłok wykazała śmierć gwałtowną, wskutek wypicia żrącej cieczy, zarazem jednak nie znaleziono tam ani ługu kaustycznego ani kwasu. Co takiego zatem wypił Gustaw?
Zbadaniem wnętrzności zmarłego zajął się świetny chemik
Jan Servais Stas, znany z badań nad ustaleniem względnej masy atomowej. On także nie znalazł ani potażu ani kwasu, co jednak zastanowiło go, to że oprócz silnej woni octu wyczuł w preparatach wyraźny zapach cygar. On sam nie znosił tej używki, zakazywał nawet asystentom palenia przed pracą, gdyż byli wówczas przesiąknięci nieznośnym zapachem. Czy zmarły palił cygara? - zapytał policję. Ależ skąd, miał za słabe płuca.
Czy zatem - zastanowił się Stas - śmierć mogło wywołać otrucie wyciągiem tytoniowym? Jeśli nawet tak było, stanowiło kłopot udowodnienie tego przed sądem. Dotychczas bowiem nikt nie opracował metody, mogącej niezbicie dowieść otrucia alkaloidami roślinnymi. Zatem należało taką metodę opracować...
Tytoń szlachetny to silnie rosnąca, jednoroczna roślina zielna z rodziny psiankowatych. W dobrych warunkach dorasta do trzech metrów, szeroko rozkładając duże, gęsto owłosione liście. Pochodzi prawdopodobnie z Andów, i stanowi hybrydę dwóch innych dzikich gatunków w tym machorki, ślady uprawy w tamtym regionie pochodzą z aż XV wieku p.n.e.[1] Ślady użytku w medycynie Indian ameryki północnej pochodzą z pierwszych wieków naszej ery. Wyciągi rośliny używane były do opatrywania ran i jako środek przeciwbólowy, wraz z szałwią na kaszel i choroby płuc. Tytoń palony w drewnianych fajkach, często z dodatkiem innych ziół, był środkiem mogącym zarówno oczyszczać myśli jak i prowadzić do odurzenia z halucynacjami, z tego też powodu uważany był za dar bogów i używany w magicznych i szamańskich rytuałach. Słynna "fajka pokoju" stanowi najbardziej znany przykład; jej palenie było zarówno symbolem zawarcia przymierza jak i sposobem odprawiania modlitw, które wraz z dymem miały unieść się do nieba. Ceremonia palenia była traktowana jak rytuał religijny, a roślina jak świętość, z tego też powodu nadużywanie tytoniu było powszechnie potępiane.
Po przybyciu pierwszych Europejczyków, zostali oni poczęstowani tytoniem. Stanowił on też prezent pojednawczy a nawet walutę. Oczywiście biały człowiek nie specjalnie przejmował się rytuałami i obyczajami tubylców, toteż tytoń zaczęto stosować jako codzienną używkę. Za pierwszego nałogowego palacza uważany jest powszechnie członek pierwszej wyprawy
Rodrigo de Jerez, który spotkał się na Kubie ze zwyczajem palenia zwitków liści. Tak sobie to przypodobał, że zebrawszy odpowiedni zapas, praktykował palenie w rodzinnym miasteczku Ayamonte a nawet zachęcał sąsiadów.
Nie skończyło się to dla niego zbyt dobrze - hiszpańskiej inkwizycji doniesiono wkrótce, że Rodrigo praktykuje pogański zwyczaj, zaś po krótkim procesie uznano, iż tylko diabeł mógł sprawić, że człowiek zyskuje siły po wdychaniu dymu i chce otaczać się jego niemiłym zapachem, toteż skazano go na pokutę, siedem lat więzienia i tym samym przymusowy odwyk.
Koloniści szybko polubili nową używkę. Pierwszym który wysyłał tytoń do Europy był Diego Kolumb, najstarszy syn Krzysztofa. Potem jednak szerzej znanym propagatorem stał się francuski dyplomata
Jean Nicot, który spotkał się z używką jako ambasador w Lizbonie. Uważał go za wartościowe zioło lecznicze, zażywane w formie naparów, przez żucie lub wziewnie po roztarciu na proszek, czyli tabakę. Opublikował nawet pracę w której polecał zażywanie jako lek na najrozmaitsze choroby, i zapewne skończyłoby się na paru traktatach, gdy by nie to, że Katarzyna Medycejska doznawała w tym okresie silnych migren.
Nicot polecił jej tabakę, co najwyraźniej poskutkowało a królowa polubiła nową roślinę. Za jej przykładem tabakę zażywali ludzie dworu aż wreszcie stała się popularną używką wśród arystokratycznej młodzieży.
O Nicot'cie pamiętano jeszcze wówczas, gdy ustalano pozycję systematyczną rośliny. Nazwano ją
Nicotiana tabacum właśnie dla utrwalenia tego pierwszego propagatora. Nazwa tabacum wywodzi się z określenia używanego przez tubylców,
tabaco lub
tavaco, które jednak w rzeczywistości oznaczało trzcinową rurę w której palono liście wciągając dym nosem, a nie samo zioło.
Niespełna wiek po wprowadzeniu do Europy, tytoń miał status panaceum. Leczono nim krosty, bielactwo, czyraki, owrzodzenia a nawet raka, polecano na długowieczność, niestrawność, osłabienie, zimnicę i co tylko przyszło medykom do głowy. W czasie epidemii był uważany za środek zapobiegawczy. Ponieważ w tym czasie za przyczynę epidemii uważano lotne wapory i złe powietrze, wydawało się że wystarczy zabić czymś niemiłe zapachy, a choroba przestanie się roznosić. Idąc tym tropem władze Londynu podczas epidemii w 1665 roku rozdawały tytoń dzieciom uczącym się w szkołach, oraz biedocie, polecając palić w pomieszczeniach.
Zarazem jednak tytoń był lekiem mało bezpiecznym, w nadmiarze wywoływał wymioty, kołatanie serca i zaburzenia oddychania. Czasem nadużycie soku lub wywaru kończyło się zatruciem, co z uwagi na dużą zmienność zawartości substancji zależnie od rośliny było trudne do uniknięcia. Z drugiej strony ówczesna medycyna chętnie stosowała takie środki jak arszenik czy strychnina, więc na tym tle tytoń nie wypadał jeszcze tak źle.
Niezwykłą popularność zyskał sobie tytoń w bardzo nietypowym zastosowaniu, które przebija nawet najdziwniejsze pomysły dzisiejszej medycyny alternatywnej. Była to lewatywa tytoniowym dymem.
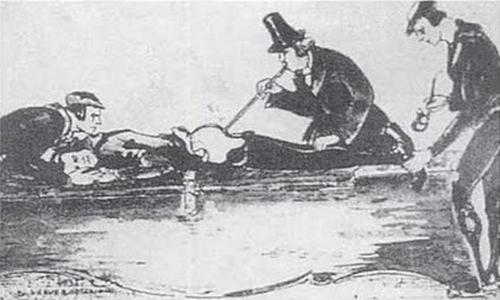
Medycyna w tym czasie nie była pewna co właściwie jest przyczyną śmierci w takich przypadkach jak utonięcie lub uduszenie. Teoria humoralna mówiła, że utonięcie powoduje powstanie w ciele nadmiaru wilgoci i wychłodzenie, z drugiej strony trudno było nie zauważyć roli zatamowania oddechu. Praktycznym rozwinięciem teorii były metody "ratowania" polegające na wlewaniu do ust alkoholu, poruszania ramionami i nogami, oraz rozcierania członków, by pobudzić krążenie i rozgrzać ciało, co można zresztą znaleźć w wielu starszych powieściach. Lewatywę dymem uważano za bardzo skuteczny sposób rozgrzewający i pobudzający, toteż zaczęto go stosować w przypadku utonięć. Wielu lekarzy stosowało zarówno wdmuchiwanie powietrza do płuc jak i dymu do odbytu, uważając za ważniejszy ten drugi zabieg. Przekonanie to doprowadziło do tego, iż na początku XIX wieku w różnych miejscach nad Tamizą umieszczono zestawy ratunkowe, zawierające niewielki miech, garść tytoniu oraz rurkę.
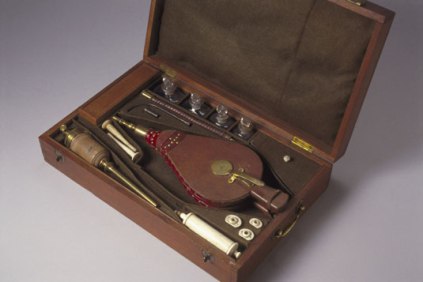
Miech mógł też służyć do respiracji płuc. Jednak już wkrótce medyczne zastosowania tytoniu zaczęły stawać się coraz mniej popularne, zwłaszcza od czasu wykazania jak bardzo toksyczny jest to środek.
Pierwsze doświadczenia nad działaniem dużych dawek przeprowadzał Benjamin Brodie, który na początku XIX wieku wykazał, że wyciąg nikotynowy może zatrzymywać akcję serca. W 1828 roku Posselt izoluje z liści oleisty składnik aktywny, nazwany nikotyną.
Nikotyna to alkaloid o wyraźnych właściwościach pobudzających, oraz stosunkowo prostej budowie, jest to pierścień pirydyny połączony z pięciokątną pirolidyną. Atom węgla przy wiązaniu łączącym ma niesymetryczne otoczenie, stanowiąc decydujące o czynności optycznej centrum setereogeniczne. Z tego też powodu możliwe stają się dwie formy różniące się konfiguracją i podobne do siebie jak lustrzane odbicia - naturalna (-)-nikotyna i syntetyczna (+)-nikotyna. Różnią się też toksycznością.
Czysta nikotyna to oleista, brązowa ciecz, mieszająca się z wodą, natomiast zapach zwykle opisywany jest jako ostry - chociaż gdy miałem ostatnio okazję powąchać czystą nikotynę w laboratorium, zdziwił mnie miękki zapach, przypominający podpieczony wafel od lodów. Działając bezpośrednio na tkanki wywołuje podrażnienia i oparzenia. Już dawka
1-1,5 mg/kg masy ciała może wywołać śmiertelne zatrucie, co oznaczałoby toksyczność równie wysoką jak cyjanek, u nałogowców występuje pewna tolerancja.
Jest aminą o słabych właściwościach zasadowych. Pod wpływem kwasów zamienia się w sól amoniową bardzo dobrze rozpuszczalną w wodzie
Zawarta w tytoniu ulatnia się podczas spalania liści, ale nie w całości lecz tylko jakieś 10%. Wdychana wchodzi w kontakt z nabłonkiem wyścielającym drogi oddechowe. I tutaj znaczenie dla wchłaniania mają własności związku - dym jest zazwyczaj kwaśny i przejściowo zakwasza śluz pokrywający nabłonek. W takich warunkach związek przechodzi w formę jonową, która słabo wchłania się do organizmu, dlatego często papierosy zawierają dodatki uwalniające amoniak alkalizujący dym i zwiększający wchłanianie. Lekko zasadowa jest w normalnych warunkach ślina, dlatego
żucie tytoniu pozwala wchłonąć znacznie więcej aktywnych substancji. W pewnych regionach działanie dodatkowo wzmacnia się, dodając do żutego tytoniu sody lub wapna (dokładnie ten sam mechanizm działa z betelem i koką, których liście były przeżuwane z wapnem).
Wciąganie
tabaki podobnie jak palenie i żucie, także opiera się na
wchłanianiu nikotyny, tyle że poprzez śluzówkę nosa, i to właśnie ten
efekt ma działanie pobudzające, nie zaś samo kichanie. W formie tabaki
tytoń przywędrował do Polski. Początkowo były to tabaki na pół
chałupnicze, ucierane w nieemaliowanych garnkach glinianych czy
donicach, często z dodatkiem popiołu, aby bardziej w nosie kręciło. Za
króla Augusta znaną producentką tabaki stała się niejaka
Syrakuzana,
Włoszka urabiająca tabakę w formie groszków rozcieranych w palcach.
Tytoń był zaprawiany lawendą lub skórką pomarańczy, a dla większej
ostrości dodawano do niego siarczanu cynku lub żelaza. Jej tabaka
zyskała taką popularność, że zaczęła być podrabiana w innych regionach.
Złośliwi przekręcali jej imię na Srajkoziana. Bywało, że dla większego szczypania dodawano do tabaki pieprzu, tartych cegieł czy nawet soli.[2]
W Krajach Skandynawskich popularnym sposobem zażywania tytoniu jest
snus, czyli torebeczka z tytoniem wkładana pod wargę i ssana.
Ostatecznie najbardziej popularnym sposobem zażycia tytoniu, jest jego palenie. Początkowo europejczycy naśladowali tubylców, paląc tytoń w trzcinowych rurach i wdychając dym nosem. Potem popularniejsze stały się
fajki a dym zaczęto wdychać ustami. Poza fajkami zwykłymi i wodnymi znano właściwie tylko
cygara robione ze skręconych liści, zawierające duża ilość rośliny. Dopiero potem zaczęto produkować mniejsze i wygodniejsze
cygaretki, zawierające wewnątrz masę z pokruszonych liści zawiniętych w pojedynczy liść.
To co znamy dziś jako papierosy jest wynalazkiem względnie młodym - pomysł cygaretek zawijanych w cienki papier wprowadził w 1880 roku
Albert Bonsack. Wynalazku wraz ze specjalną maszynką do zawijania początkowo nie chciano kupować, uważając że "papierowa cygaretka" jest gorsza w smaku i pewnie robi się ją ze zmiotek po cygarach. Pewien wpływ miały tu też obawy ze strony producentów cygar - glizownica pozwalała w krótkim czasie wyprodukować tyle papierosów, ile zakładom z wieloma robotnikami zajmowało tygodnie. Wynalazca wszedł więc w odpowiednią spółkę i zaczął produkować papierosy, które za sprawą niższej ceny i poręcznych rozmiarów szybko zyskały popularność na całym świecie. W Polsce pojawiły się na przełomie XIX i XX wieku.
Upowszechnienie papierosów spowodowało też jego umasowienie, co szybko zaczęło wywoływać kłopoty. Już w XIX wieku lekarze wyrażali obawy o wpływ palenia na zdrowie. Było wiadomo że wdychanie dymu przez węglarzy i kominiarzy szkodzi na płuca i że wśród ludzi tych częstszy jest rak płuca, dlatego w 1912 roku
dr Azaak Adler ogłosił, że papenie tytoniu działa podobnie. Jednak badania polegające na obserwacji że wielu chorych na raka paliło, były niedostateczne - w końcu paliła duża część społeczeństwa.
Po zakończeniu I wojny światowej zaobserwowano gwałtowny wzrost zachorowań na nowotwory płuc, krtani i języka, co w latach 30. poważnie zaniepokoiło lekarzy. Dziwne jest w tej sytuacji zignorowanie doniesienia niemieckiego lekarza
Fritza Lickinta, który najpierw w 1925 roku zwrócił na częstsze nowotwory żoładka u palaczy, a potem w 1929 wydał obszerną pracę statystycznie udowadniającą związek palenia z rakiem płuc.
Do zignorowania Lickinta częściowo przyczyniła się propaganda niemieckich firm tytoniowych, a częściowo jego poglądy polityczne - był socjaldemokratą, co wobec rosnącego w siłę ruchu nazistowskiego było niepożądane. W 1934 stracił pracę, a potem został wcielony do wojska jako lekarz frontowy, gdzie przeżył całą wojnę. Ironią losu było to, że jego wyniki stały się potem podstawą dla nazistowskiej kampanii antynikotynowej.

Stanowiący centrum nazistowskiej ideologii plan przebudowy społeczeństwa i wykształcenia idealnego narodu, obejmował też eliminację czynników osłabiających silę i morale. Za jeden z nich uznano szkodliwe używki, a w szczególności palenie papierosów. W dodatku wczesne badania wskazywały na zmniejszenie płodności za
sprawą większej ilości poronień i wad u dzieci matek palących - a
przecież do budowy nowego społeczeństwa potrzebne były w pełni płodne,
zdrowe matki. Dlatego w latach 30. doprowadziło to do zakazów palenia w tramwajach i miejscach publicznych, znane stało się też wtedy pojęcie "palenia biernego".
Nazistowscy badacze opublikowali w latach
1939-45 szereg obszernych badań wskazujących na szkodliwość palenia, w tym badań z grupą porównawczą, spełniających wszelkie wymogi statystyki. Za najważniejszy skutek palenia uznano wtedy choroby serca. Pod koniec wojny pojawiła się nawet praca opisująca wyniki autopsji kilkudziesięciu żołnierzy, którzy zmarli z powodu zawału - większość była palaczami i miała zniszczone płuca. Sam Hitler był przeciwnikiem palenia - uważał je za objaw dekadentyzmu, oraz "zemstę czerwonych", a także osłabiający nałóg osłabiający tężyznę fizyczną. Namawiał współpracowników do rzucenia nałogu i zawsze irytował go zwyczaj Goeringa do palenia w pomieszczeniach. Miał plany aby po wygranej wojnie zakazać palenia wszędzie.
Kampania antynikotynowa była brudną kampanią - obok uzasadnień ideologicznych chwytała się kojarzenia palenia z "żydowskimi zwyczajami", pisano też że to brudny murzyński zwyczaj niegodny cywilizowanego, białego aryjczyka. Do kampanii włączył się niemiecki kościół Adwentystów Dnia Siódmego. Doszło nawet do wydawania czasopisma z bajkami i śmiesznymi obrazkami o szkodliwych skutkach nikotynizmu.
Kampania była jednak mało skuteczna - aż do rozpoczęcia wojny konsumpcja papierosów rosła bardzo szybko. W czasie wojny przyhamowała głównie z powodu niedoboru surowca na rynku, mimo wzrostu ilości palaczy.
Zaraz zaraz - skoro już wtedy, w tych wojennych czasach ukazały się szczegółowe, dobrze wykonane prace o szkodliwości palenia, to dlaczego po wojnie papierosy nadal były uważane za nieszkodliwe? Bo tamte prace były nazistowskie.
Dlatego też świat musiał poczekać aż do roku 1950 gdy brytyjski lekarz
Richard Doll opublikował swój raport, wykazujący związek palenia z rakiem płuc. Trzy lata później głośna stała się praca opisująca powstawanie raka skóry u myszy posmarowanych smołą tytoniową.
Dziś szkodliwy wpływ papierosów jest już dobrze udowodniony. Główne znaczenie mają tu rakotwórcze produkty częściowej pirolizy tytoniowych okruchów, takie jak arkoleina czy wielopierścieniowe węglowodory aromatyczne, częściowo metale ciężkie jak ołów i kadm zawarte w nawozach a w pewnym stopniu też promieniotwórczy polon gromadzący się na powierzchni liści. Na zdrowie palaczy wpływ ma też tlenek węgla zawarty w dymie.
Po wchłonięciu bardzo szybko zostaje rozprowadzona po organizmie, docierając do mózgu gdzie wywiera właściwe działanie, stanowiąc inhibitor receptora acetylocholinowego.
Komórki nerwowe utrzymują stale pewną nierównowagę ilości jonów między wnętrzem i zewnętrzem. Aktywny transport jonów doprowadza do sytuacji, gdy po wewnętrznej stronie błony komórkowej jest więcej anionów niż w płynie na zewnątrz. W efekcie powstaje niewielki potencjał elektryczny ok. -70 mV. Jego rozładowanie zachodzące poprzez otworzenie kanałów jonowych w błonie i pozwolenie jonom na wpływanie do komórki, wywołuje miejscowe powstanie przeciwnego potencjału o wielkości ok. +30 mV. Ta zmiana potencjału rozchodzi się od neuronu do neuronu, tworząc impuls nerwowy.
W tworzeniu tej elektrycznej nierównowagi udział biorą głownie jony potasu i sodu, których stężenia są sztucznie zmieniane przy pomocy kanałów jonowych - tworów przechodzących przez błonę i wyrzucających na zewnątrz sód i wciągających do środka potas.
Kanały te mogą otwierać się aby przepuścić jony w którąś stronę, jeśli jest to organizmowi potrzebne, co sygnalizują odpowiednie neuroprzekaźniki. Takim kanałem są między innymi receptory acetylocholinowe zlokalizowane w błonach neuronów. Aby kanał się otworzył, organizm musi wydzielić agonistę, czyli substancję która wiążąc się z receptorem otworzy kanał. Taki uniwersalnym agonistą jest acetylocholina, związek będący prostą, czwartorzędową aminą. Identyczne działanie może mieć jednak wiele innych substancji będących aminami, w tym nikotyna.
Otworzenie kanału jonowego zmienia polaryzację błony, co wywołuje krótkotrwałe pobudzenie układu nerwowego. Dlatego substancje będące agonistami tego receptora będą miały działanie pobudzające. W przypadku nikotyny szczególnie chętnie łączy się ona z receptorami komórek nerwowych w nadnerczach, wywołując uwalnianie adrenaliny. Uaktywnia też wydzielanie dopaminy, stąd poprawa samopoczucia.
Ale jak to już wielokrotnie wykazywałem, co za dużo, to nie zdrowo. Kanały jonowe nie powinny pozostawać otwarte zbyt długo, dlatego cholina i nikotyna zwykle dosyć szybko odłączają się od receptora. Jednak przy dużych dawkach, zanim komórka powróci do pierwotnej polaryzacji, receptor jest znów otwierany. Przedłużona depolaryzacja błony wywołuje ostatecznie efekt odwrotny do pierwotnego - aktywność układu nerwowego zmniejsza się.
Objawy zatrucia nikotyną są zazwyczaj dość charakterystyczne. Najpierw następuje faza nadmiernego pobudzenia, co daje takie objawy jak nadmierne pocenie i ślinienie, podwyższone ciśnienie, szybkie bicie serca, drżenie przechodzące w drgawki, bóle brzucha i głowy, wymioty itp. Po tej fazie następuje druga, związana z działaniem hamującym nadmiernych dawek. Następuje gwałtowny spadek ciśnienia, niedowłady i duszności przechodzące w ustanie oddechu wskutek osłabienia mięśni oddechowych. Przyczyną zgonu zwykle jest uduszenie lub ustanie czynności serca.
Dawka śmiertelna to ok. 500 mg nikotyny, o jest raczej trudne do osiągnięcia przez samo palenie papierosów. Teoretycznie możliwe jest przy kombinacji palenia, gum nikotynowych i plastrów. Możliwe jest też niesmiertelne zatrucie w wyniku ssania snusów i tego typu pakietów nasączonych łatwo wchłanialnymi solami nikotyny. Zanotowano przypadek śmierci dwóch nastolatków, którzy wdychali dym z rury napełnionej tytoniem, którzy za pomocą tej zaimprowizowanej fajki chcieli wywołać u siebie halucynacje, ale to ekstremum.
Większość notowanych zatruć wynikała więc raczej z kontaktu z środkami owadobójczymi na bazie nikotyny. Alkaloid jest dla owadów znacznie bardziej toksyczny niż dla ludzi, i dlatego sok tytoniu, wyciąg wodny a nawet pył z roztartych liści już od wieków były używane do zwalczania szkodników. Aż do lat 50. insektycydy stanowiły drugie najważniejsze zastosowanie tytoniu. Obecnie jednak wycofuje się je ze względu na zbyt mało specyficzne działanie, nikotyna i jej pochodne zabijają bowiem też zwierzęta, no i oczywiście są niebezpieczne dla ludzi. Pochodne z grupy neonikotynidów były uważane za bezpieczną alternatywę ze względu na niską toksyczność wśród ssaków, jednak powiązano je ze zjawiskiem masowego ginięcia pszczół i dlatego są wycofywane.

Jest jednak jeszcze jedno źródło nikotyny, coraz popularniejsze i tym samym niebezpieczne - liquid, czyli płynny wkład do papierosów elektronicznych. Jest to zazwyczaj roztwór nikotyny w glicerynie lub glikolu propylenowym z dodatkami zapachowymi i smakowymi. stężenia nikotyny w takich płynach mogą być dosyć duże, dlatego notowano już zatrucia związane z nieostrożnym obchodzeniem się. Niekiedy wystarcza rozlanie wkładu na dłonie, ręce czy tułowie a także na ubranie mające potem dłuższy kontakt ze skórą, bowiem nikotyna wchłania się przez skórę. Inne przypadki dotyczyły zwilżenia ustnika, czy jedzenia dłońmi na które wcześniej wylał się liquid.
Bardzo wiele zatruć dotyczy dzieci, które liżą niedokręcone buteleczki, przyciągnięte słodkim, owocowym zapachem, lub wypijają płyn z otwartych pojemników, albo wdychają mgiełkę z pozostawionych włączonych urządzeń.
Wzrost liczby takich przypadków jest dramatyczny - już w tym roku w Stanach Zjednoczonych zdarzyło się 2400 zatruć z tego powodu, z czego ponad połowa dotyczyła dzieci, część z nich wywołała zgon.[3] Niektóre z tych zatruć dotyczyły nastolatków zaprawiających sobie drinki dla wzmocnienia, inne dotyczyły dorosłych smarujących się płynem w zastępstwie plastra nikotynowego.
W przypadku rozlania liquida na skórę, powinno się go szybko zetrzeć, a skórę umyć mydłem. Podobni powinno się postępować z powierzchniami na które wylał się lub kapnął płyn. W przypadku ubrania powinno się je zdjąć, nawet jeśli wydaje się że plama wyschła. Po przeczytaniu tego fragmentu powinniście się już orientować, że pojemniczków nie powinno się przechowywać w zasięgu dzieci i zwierząt domowych bo może to być dla nich śmiertelnie niebezpieczne.
W przypadku połknięcia nikotyny, jedną z metod leczenia może być podanie węgla aktywnego, zmniejszającego wchłanianie. Leczenie szpitalne zatruć polega głównie na łagodzeniu objawów - w fazie nadmiernego pobudzenia środkami uspokajającymi a w fazie osłabienia podawaniem atropiny, regulacją ciśnienia i wspomaganiem oddechu. Przy zatruciu ostrym z zatrzymaniem oddechu ważna jest sztuczna wentylacja, wówczas bowiem większą szkodę wywołuje niedotlenienie niż samo zatrucie. Zazwyczaj przy takim wspomaganiu objawy ustępują po paru dniach w miarę metabolicznego przerobu nikotyny, i nie pozostawiają długotrwałych następstw.
A co tam u hrabiostwa? Podczas śledztwa zwrócono uwagę na sprzęt laboratoryjny w domu hrabiego. Podobno w ostatnim czasie zainteresował się chemią. W dodatku znaleziono u niego książkę na temat trujących roślin, w tym także o właściwościach tytoniu.
Służba opowiedziała sędziemu, że w lato Bocarme zamówił duże ilości ciętego tytoniu, rzekomo na zapas do skręcania cygaretek, choć nie widziano potem aby tak często palił. Jeśli połączyć to z informacją o zakopanych w kącie ogrodu zdechłych nagle okolicznych psach i kotach, cała historia zaczyna wyglądać jasno. Hrabia kupił tytoń, z niego poprzez gotowanie z octem otrzymał wyciąg, który zagęścił; z wyciągu wyizolował czystą nikotynę, której działanie testował na zwierzętach. Wyglądało to zatem na działania planowane już od dawna.
Jednak dla sądu pomiędzy stwierdzeniem, że oskarżony mógł zdobyć niebezpieczną truciznę, a stwierdzeniem że to nią otruto Gustawa, zachodziła istotna różnica. Należało zatem tą truciznę w ciele zmarłego wykryć.
Gdy Jan Servais Stas zastanawiał się nad wyizolowaniem trucizny z tkanek zabitego, medycyna sądowa nie dawała na to zbyt wielkich nadziei. Były już znane techniki wykrywania we wnętrznościach trucizn nieorganicznych, zazwyczaj w tym celu próbkę spalano lub rozpuszczano w mocnym kwasie, który niszczył substancje organiczne pozostawiając sole trującego metalu. Nikotyna jest jednak trucizną organiczną, i nie można było jej niszczyć. Dlatego wpadł na inny pomysł.
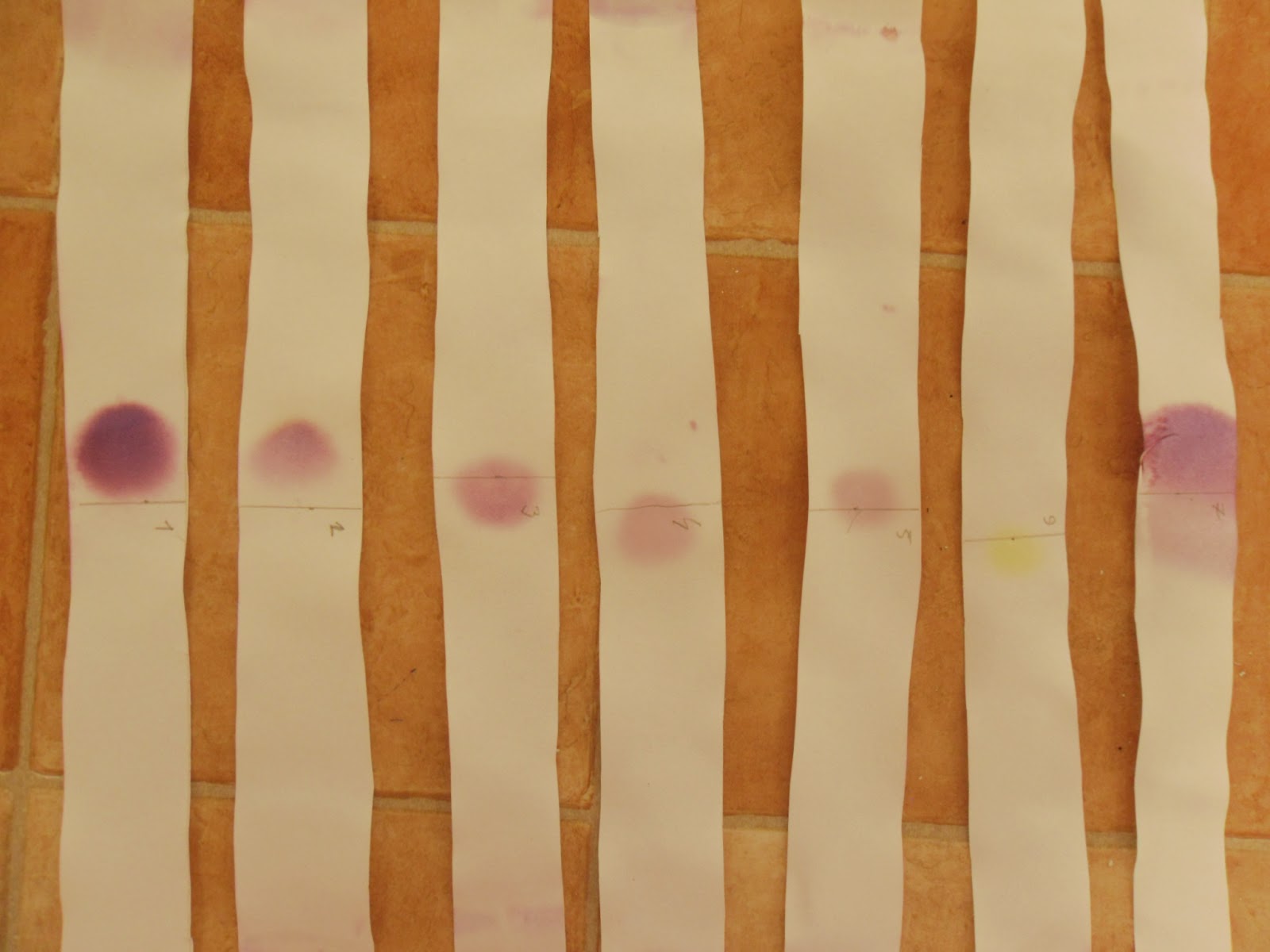
Pobrał część żołądka zabitego i wytrawiał w rozcieńczonym kwasie, przeprowadzając alkaloid w rozpuszczalne sole i tym samym wypłukując go z tkanki. Otrzymany płyn potraktował zasadą, która rozpuściła resztki białek i przeprowadził nikotynę do wolnej postaci. Na koniec zagęścił powoli odparowując.Gdy Stas otrzymał już płyn z roztworzenia żołądka, wykorzystał znaną właściwość nikotyny do rozpuszczania się w eterze. Ekstrahował mieszaninę eterem, który odparowywał. Na dnie pozostała mu już tylko oleista ciecz o charakterystycznym zapachu, którą poddał próbom charakterystycznym z kwasami, potwierdzając, że reaguje identycznie jak nikotyna. A skoro tak, to musiała być nikotyna.
[4]
Tym samym można było potwierdzić, iż zabójstwa dokonano tym związkiem. Gdy to nastąpiło, rozpoczął się proces, który ze względu na stan społeczny oskarżonych wzbudził zainteresowanie w całej Europie.
Początkowo hrabiostwo wszystkiemu zaprzeczali, jednak po rozpoczęciu procesu zgłosił się profesor Loppens, którego przez kilka miesięcy pewien człowiek wypytywał o technologię wyodrębniania nikotyny, tłumacząc mu, iż będąc w Ameryce widział, jak Indianie zatruwają strzały sokami pewnych roślin, że zaś ma za oceanem rodzinę do której zamierza znów przyjechać, pragnie zdobyć wiedzę o takich truciznach. Człowiek ten, posługujący się nazwiskiem Bernard, raz nawet odwiedził go pokazując próbki ekstraktów i informując, że sprawdzał je już na zwierzętach z piorunującym skutkiem.
Śledczy przeprowadzili małą konfrontację, dając profesorowi okazję zobaczenia hrabiego. Był to dokładnie ten sam człowiek. Profesor zachował listy, napisane jak oceniono ręką hrabiny. Mając w ręku taki dowód, śledczy przycisnął hrabinę, grożąc że może zostać uznana za morderczynię. Przestraszona Lidia przyznała - tak, Gustaw został zabity. Ale sprawcą był mąż Hippolit. Podszedł od tyłu do jej brata i trzymając jedną ręką za głowę, włożył mu dwa palce głęboko do ust, wlewając truciznę między rozwarte zęby. Gdy ciałem wstrząsnęły konwulsję trzymał go, dopóki ten nie zwiotczał.
Podczas procesu urządzono konfrontację małżonków. Lidia oskarżała męża o maltretowanie i przymuszanie do zbrodniczego planu, natomiast hrabia zbywał te słowa uśmiechem. Przez większość procesu zachowywał się swobodnie, uważając że dowody są zbyt słabe. Dwuznacznie chwalił się wielką znajomością trucizn i dużym wkładem w toksykologię.
Nie mogąc zaprzeczyć, że wyrabiał nikotynę i że od nikotyny zginął Gustaw, twierdził że zmarły wypił fiolkę z nikotyną stojącą na kredensie, gdy hrabiostwo udali się na chwilkę do kuchni po drugie danie. Gdy nie dano temu wiary, zmienił punkt obrony - to Lidia dała truciznę bratu, mówiąc mu że to koniak, a nawet rozlała jej nieco na suknie i dłonie. Dlatego właśnie, jak widziała służba, zaraz po otworzeniu sali jadalnej poszła umyć ręce, dlatego kazała ubrania swoje i męża uprać jeszcze tej samej nocy, i dlatego wreszcie wrzuciła do pieca kule zmarłego brata.
Brat miał odjeżdżać i poprosił o szklankę koniaku na odjezdne. Lidia wzięła dwa kieliszki i postawiła je na kredensie, gdzie napełniła je z butelki, tak że nie widzieli co nalewa. Dała kieliszki obydwu, Gustaw wychylił swój duszkiem, mąż zaledwie przytknął do ust nim poznał po zapachu nikotynę. Gdy Gustaw poznał, że to co wypił było trucizną, zaczął krzyczeć. Wtedy hrabia zamknął mu dłonią usta aby nie wywołał skandalu i został ugryziony. Ponieważ nieco trucizny znalazło się na jego języku, stracił na chwilkę przytomność i upadł, co tłumaczy stan jego ubrania, ponadto uderzył się w kredens co tłumaczy ranę na czole którą wzięto za ślad paznokci zabitego. Co do motywów żony, najpierw niejednoznacznie dawał do zrozumienia, że między rodzeństwem panowała głęboka nienawiść, lecz zaraz potem twierdził, że nalanie do kieliszków trucizny było nieszczęśliwym zbiegiem okoliczności - butelka z alkoholem stała niedaleko butelki z trucizną i Lidia wzięła nie tą co trzeba.
Obawiając się, że nikt nie da im wiary, uznał że trzeba ukryć to zdarzenie. A dlaczego teraz żona go oskarża? Bo ona ma dużą skłonność do kłamania, pisze te romanse i ciągle coś zmyśla.
Proces przeciągał się. Powoływano coraz to nowych świadków, roztrząsano kwestie gdzie stała butelka, czy podłoga była skrobana, czy wiemy jak działa nikotyna czy też jest to rzecz niepewna, lecz ostatecznie 14 czerwca ogłoszono wyrok: Hippolit Visart Hrabia de Bocarme zostaje uznany winnym zabójstwa Gustawa Fougnies. Lidia Visart, siostra Gustawa, zostaje uniewinniona.
W późniejszym czasie wątpliwości wywoływało uniewinnienie hrabiny, która czy z własnej woli czy przez przymuszenie musiała pomagać w przygotowaniach. Ponadto wskazywano, że podczas wlewania trucizny, ofiarę musiały trzymać dwie osoby. Mimo to nie doszło do rewizji procesu.
Hrabia Hippolit został zgilotynowany 19 lipca 1851 roku.
-------
*
Zastosowania medyczne tytoniu w historii
* Gazeta Warszawska nr. 145. 2 czerwca 1851 EBUW
* Gazeta Warszawska nr. 147, 4 czerwca 1851 EBUW
* Gazeta Warszawska nr. 155, 14 czerwca 1851 EBUW
* Gazeta Warszawska nr. 171. 4 lipca 1851 EBUW
* http://murderpedia.org/male.B/b/bocarme.htm
* http://en.wikipedia.org/wiki/Tobacco
* http://en.wikipedia.org/wiki/History_of_tobacco
* http://en.wikipedia.org/wiki/Rodrigo_de_Jerez
* http://en.wikipedia.org/wiki/Tobacco_smoke_enema
* http://en.wikipedia.org/wiki/Anti-tobacco_movement_in_Nazi_Germany
* http://en.wikipedia.org/wiki/Nicotiana_tabacum
* http://en.wikipedia.org/wiki/Nicotine
* http://en.wikipedia.org/wiki/Nicotine_poisoning
* http://en.wikipedia.org/wiki/Nicotinic_acetylcholine_receptor
* http://en.wikipedia.org/wiki/Ligand-gated_ion_channel
[1] http://archaeology.about.com/od/tterms/qt/Tobacco-History.htm
[2] http://staropolscy.pl/jedrzej-kitowicz/opis-obyczajow-za-panowania-augusta-iii/o-tabace-i-wloszce-syrakuzanie-nazywanej
[3] http://www.cbsnews.com/news/sharp-rise-in-liquid-nicotine-poisonings-in-children/
[4]
Dodatek do "Chemii Policyjno Prawnej" Warszawa 1854, EBUW

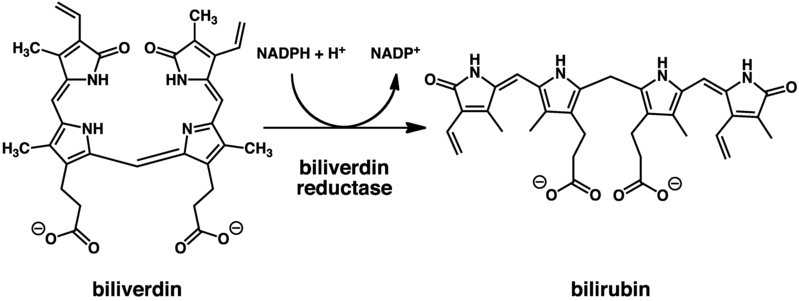
.jpg/800px-Black_eye_(3).jpg)





.jpg)