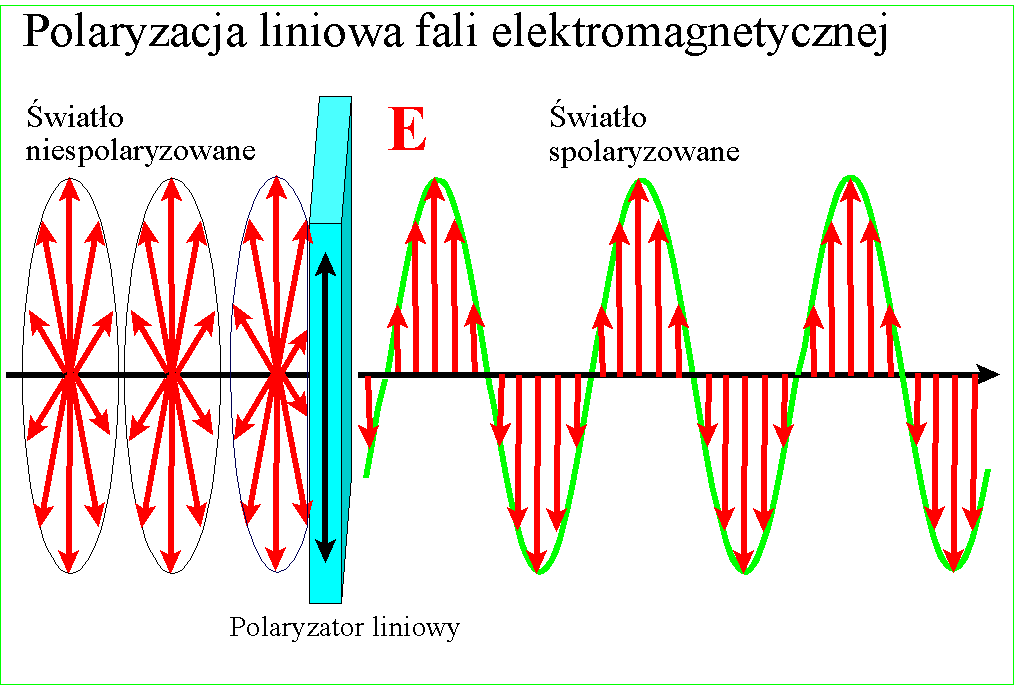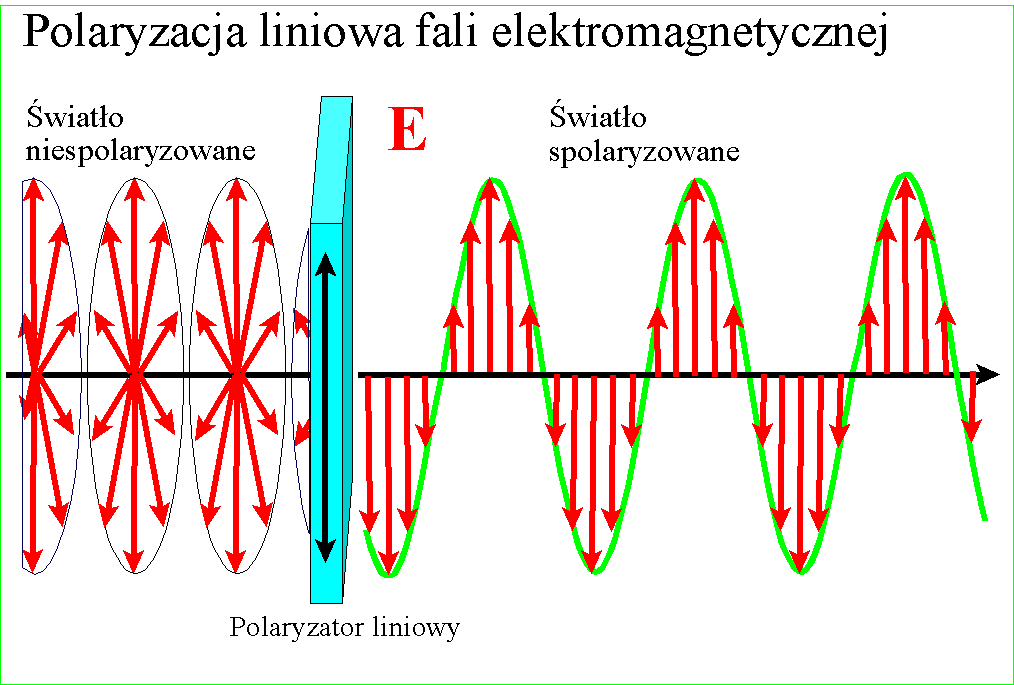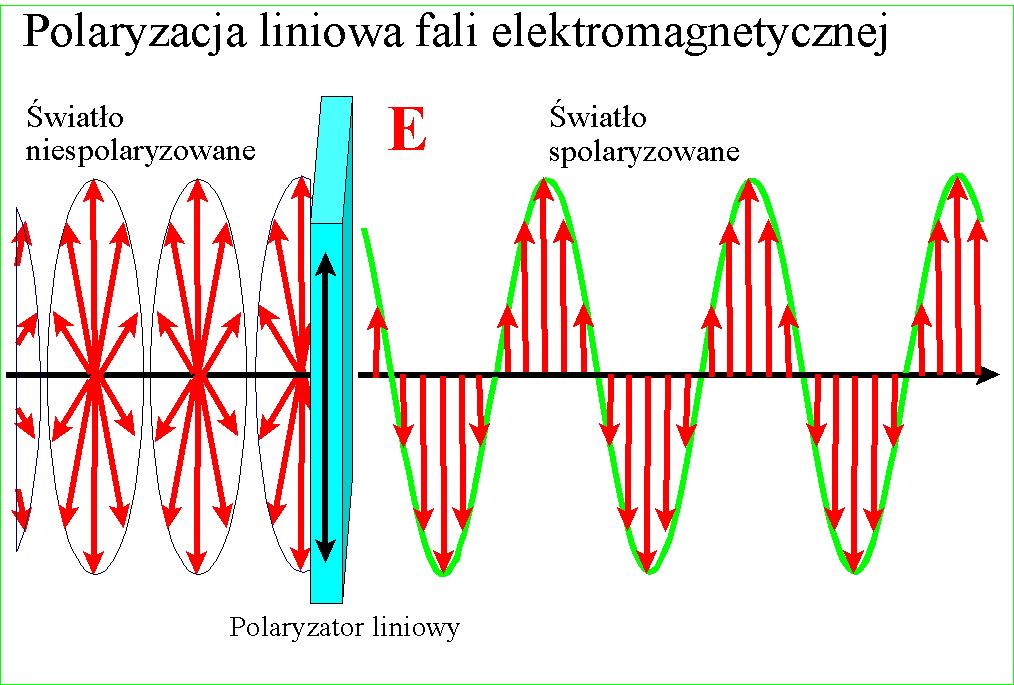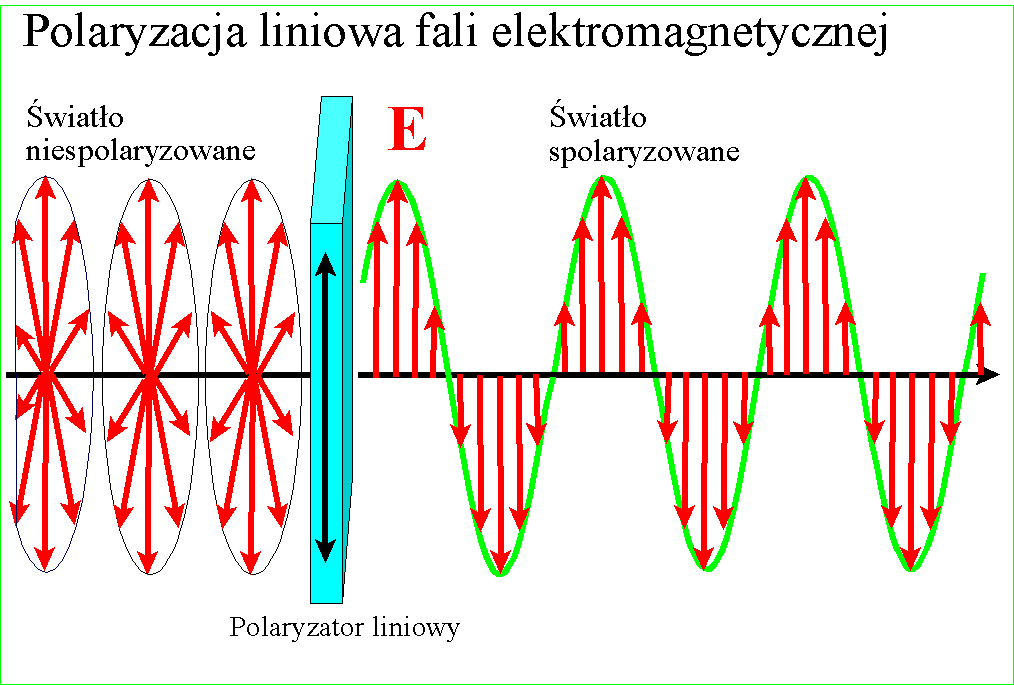
Nowa metoda wykrywania pestycydów w jedzeniu:
Problemem badań analitycznych jest oddzielenie badanych substancji od wszystkiego innego w próbce. Rozdział i przygotowanie oczyszczonej frakcji bardzo spowalniają samą analizę. Badacze z Singapuru zaproponowali ostatnio szybką metodę rozdziału do badania żywności - do rozmiksowanej próbki warzyw dodaje się mikrocząstki pochłaniające substancje podobne do pozostałości pestycydów. Cząstki zawierają wewnątrz magnetyczny tlenek żelaza, więc po pewnym czasie wystarczy zanurzyć w próbce magnes, aby oddzielić je od roztworu. Potem cząstki przemywa się rozpuszczalnikiem zmywając z nich pochłonięte substancje a otrzymany roztwór trafia do analizy. Magnetyczne cząstki można potem użyć jeszcze 30 razy.
W zasadzie jest to modyfikacja metody ekstrakcji do fazy stałej (SPE) o której pisałem przy okazji analizy zawartości kofeiny w kawie.[1]
Lustrzany świat hormonów
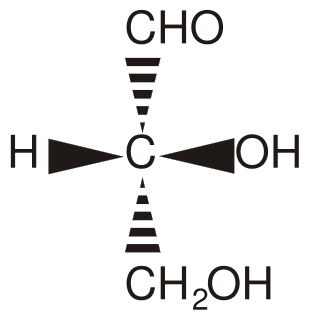
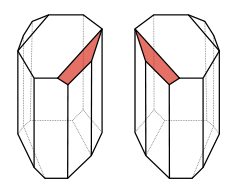
Na blogu kilkakrotnie wspominałem już o chiralnych cząsteczkach związków chemicznych. Chiralność to sytuacja, gdy cząsteczka nie posiada elementów symetrii takich jak środek czy płaszczyzna inwersyjna. Geometria ta powoduje, że możliwe stają się dwa izomery tej cząsteczki, wyglądające jak lustrzane odbicia, które nie nakładają się na siebie. Podobny kształt mają nasze dłonie. Lustrzane odbicie jednej wygląda jak druga, ale kształt jednej dłoni nie nałoży się dokładnie na kształt drugiej, choćbyśmy je nie wiem jak wykręcali. Stąd też bierze się nazwa - chiralność od greckiego słowa chira, czyli ręka.
Lustrzane izomery tej samej cząsteczki mają takie same właściwości fizyczne i chemiczne, ale różnie oddziałują z innymi chiralnymi cząsteczkami. Najlepszym tego świadectwem jest różne działanie biologiczne izomerów geometrycznych leków. My sami zbudowani jesteśmy z chiralnych białek, a te z chiralnych aminokwasów. Wszystkie aminokwasy budulcowe w organizmie stanowią izomer L.
Ponieważ jednak umiemy już dziś syntetyzować nienaturalne izomery aminokwasów, czyli izomery D, oraz umiemy sztucznie syntezować białka o zadanej kolejności aminokwasów, rodzi się oczywiste pytanie - jak mogłyby działać na organizm lustrzane izomery całych białek?
 |
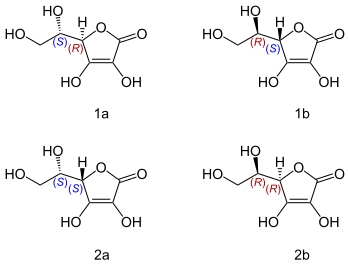
| Przykładowe izomery prostego tripeptydu. Credit: Pablo Gainza |
Badacze z uniwersytetu w Toronto zainteresowali się tym tematem podczas poszukiwań nowych leków. W niektórych chorobach stosowanymi lekami są krótkie białka, peptydy, mające pełnić odpowiednie funkcje biologiczne, zastępować enzymy których w organizmie brakuje, lub blokować te, których jest nadmiar. W leczeniu cukrzycy używane są na przykład mimetyki GLP-1, naśladujące białko które aktywizuje enzymy inkretyny, co wpływa na obniżenie poziomu cukru we krwi.
Ich stosowanie wiąże się jednak z licznymi problemami - muszą być wstrzykiwane domięśniowo, bo w układzie pokarmowym są trawione przez enzymy trawiące białka; we krwi krążą krótko, rozkładane przez inne enzymy, w dodatku organizm może zareagować na nie alergicznie.
Badacze przeprowadzili symulację, sprawdzając czy istnieją D-izomery krótkich peptydów, które będą tak samo dobrze aktywować GLP-1. Znaleźli pewną obiecującą cząsteczkę, którą zsyntezowano i przetestowano. Otrzymany związek wywoływał taką samą odpowiedź komórek, ale efekt utrzymywał się dłużej.
Ponieważ wszystkie białka w organizmie mają konfigurację L, to peptyd będący izomerem D będzie po pierwsze nie trawiony przez enzymy trawienne bo te, same będąc cząsteczkami chiralnymi, są przystosowane do trawienia L-peptydów, dzięki czemu lek będzie można zażywać w tabletkach; oraz po drugie D-peptyd nie będzie tak szybko eliminowany z krwioobiegu co przedłuży jego działanie i spowoduje, że potrzebne będą mniejsze dawki.
Podobny D-izomer otrzymano też dla peptydomimetyków parathormonu, które podawane są przy zaburzeniach czynności przytarczyc. [2]
Sztuczny słodzik w chemioterapii
Dokładne badania molekularne wykazały kilkanaście lat temu, że w wielu złośliwych nowotworach wyjątkowo dużą ekspresję posiada enzym anhydraza węglanowa IX, która normalnie jest wydzielana w nerkach do usuwania kwasu węglowego z krwi. Jej duża aktywność sprzyja szybkiemu rozrostowi guzów i naciekaniu na okoliczne tkanki. Zaczęto więc uzupełniać terapię o inhibitory tego enzymu, dotychczas stosowane w medycynie jako diuretyki, środki zmniejszające ciśnienie wewnątrz oka czy łagodzące objawy choroby wysokogórskiej. Wstępne wyniki testów są obiecujące i niektóre takie środki wchodzą w fazę badań klinicznych.
Inhibitory te mają jednak swoje wady, w tym skutki uboczne jak wymioty, zmęczenie, objawy grypopodobne. Wiążą się one w dużym stopniu z małą selektywnością leków. Organizm wytwarza 15 różnych anhydraz węglanowych, które pełnią różne funkcje w organizmie, tymczasem nowotwory złośliwe wykazują nadekspresję tylko jednego, anhydrazy IX. Lek, który hamuje działanie nie tylko tego jednego enzymu, ale też i kilku innych, będzie wywoływał dodatkowe niekorzystne skutki.
Zaczęto zatem szukać substancji, które byłyby inhibitorami dużo bardziej selektywnymi. Niedawno doniesiono, że dość selektywne działanie wykazuje sacharyna, pierwszy sztuczny słodzik, wprowadzony do użytku jeszcze w XIX wieku. W tym roku postanowiono przetestować kolejny słodzik - acesulfam, którego struktura jest podobna do sacharyny. Okazało się, że nie tylko wiąże się z enzymem mocniej, ale też dużo bardziej selektywnie i potencjalnie mógłby być, w odpowiednio dużych dawkach, użyty jako dodatkowy składnik terapii nowotworowych. Zastosowanie to byłoby o tyle ułatwione, że jako stary, od dawna znany związek, został przebadany na bardzo wiele sposobów, a to ułatwia rejestrację w nowym zastosowaniu (w zasadzie efekty toksykologiczne, sprawdzane w badaniach klinicznych I fazy, zostały już zbadane).
Naukowcy jednak na tym nie poprzestają, sprawdzają pochodne acefulfamu, chcąc znaleźć jeszcze lepszy związek.[3]
Zdjęcie atomu
Niedawno media obiegła wiadomość, w jakimś stopniu uzupełniająca mój artykuł o obrazowaniu atomów. Udało się przeprowadzić doświadczenie, które umożliwiło ukazanie pojedynczego atomu. Pojawiły się w związku z tym różne wątpliwości ze strony mało rozumiejących eksperyment czytelników, którzy wątpili, czy atom może odbić na tyle dużo światła aby dało się go zobaczyć.
Eksperyment jest bardzo prosty, choć nie nadaje się za bardzo jako technika obrazowania atomów, nie pozwala bowiem na uchwycenie żadnych szczegółów.

Pojedynczy jon strontu został schwytany w pułapkę pomiędzy potężnymi magnesami i między dwoma elektrodami z przyłożonym napięciem. Naładowana cząstka chce poruszać się wzdłuż linii pola elektrycznego, będąc wciąganą w obszar największego gradientu dokładnie pomiędzy elektrodami. Jednak umieszczenie jej w polu magnetycznym powoduje powstanie siły Lorentza, która zakrzywia jej tor. Jon zaczyna więc kręcić małe kółeczka. W osi pionowej utrzymuje go pole magnetyczne, a pole elektryczne nie pozwala mu uciec na boki. Odpowiednia modulacja pola magnetycznego lub elektrycznego ogranicza naładowaną cząstkę do bardzo małej przestrzeni, w obrębie której może jedynie drgać. Jest to najprostszy przypadek pułapki jonowej.
Dzięki zastosowaniu odpowiednio dobranej selekcji, poprzez dodawanie jonom na tyle małych dawek energii aby pojedynczo uciekały z pułapki, można doprowadzić do stanu, gdy zamknięty pozostanie pojedynczy jon. Jak go jednak ujawnić?
Jon strontu został oświetlony ultrafioletem o tak dobranej długości fali, że w jonie zachodziła fluorescencja, to jest pochłaniał fotony ultrafioletu po czym zaraz wypromieniowywał wchłoniętą energię, ale z przesunięciem częstotliwości, świecąc już w zakresie widzialnym. Oczywiście natężenie tego świecenia dla pojedynczej cząstki jest nikłe, jednak przy użyciu odpowiednio czułego aparatu, i długiego, 30-sekundowego czasu naświetlania, nazbierało się go dostatecznie dużo, aby na fotografii ujawnił się mały, jasny punkcik.
Czy punkt ten jest obrazem atomu? I tak i nie. Atom w pułapce zatacza pewną zamkniętą orbitę, toteż gdyby zastosować duże przybliżenie, zamiast punkcika widzielibyśmy pewnie skomplikowaną krzywą. Jest ona jednak w pułapce tak mała, że w użytej rozdzielczości nadal stanowi bezszczegółowy punkt. Uchwycony obraz jest więc raczej statystycznym położeniem atomu, który emitując fotony wykonał w ciągu tych 30 sekund ogromną ilość drgań w pułapce. Zarazem jednak na obraz składa się światło pochodzące od pojedynczego atomu, wykazując, że możliwe jest optyczne wykrycie nawet tak małej cząstki materialnej.[4]
-------------
Źródła:
[1] Xi Yu et al. Pyrethroid residue determination in organic and conventional vegetables using liquid-solid extraction coupled with magnetic solid phase extraction based on polystyrene-coated magnetic nanoparticles, Food Chemistry (2016). DOI: 10.1016/j.foodchem.2016.08.115
[2] Michael Garton el al., Method to generate highly stable D-amino acid analogs of bioactive helical peptides using a mirror image of the entire PDB," PNAS (2018). http://www.pnas.org/cgi/doi/10.1073/pnas.1711837115
[3] Akilah B. Murray et al. "Seriously Sweet": Acesulfame K Exhibits Selective Inhibition Using Alternative Binding Modes in Carbonic Anhydrase Isoforms, Journal of Medicinal Chemistry (2017). DOI: 10.1021/acs.jmedchem.7b01470
[4] https://www2.physics.ox.ac.uk/research/ion-trap-quantum-computing-group