Do laboratorium analitycznego mogą czasem przychodzić dziwne próbki. Jak choćby ostatnio - przyniesiono nam próbkę osadu z bojlera, o uderzającym zielonym kolorze, z zapytaniem czy możemy zbadać jakie pierwiastki w tym są, bo coś takiego się wytrąca. Już pierwszy rzut oka sugerował miedź:
Ale oko nie spektroskop, należało to jakoś potwierdzić. Pierwsze badanie - reakcja z kwasem solnym. Osad powoli rozpuszczał się, wydzielając bąbelki gazu. Czyli w skład osadu raczej wchodził zwykły węglanowy kamień. Roztwór po rozpuszczeniu miał słaby, zielonkawo-niebieski kolor. Co jeszcze mogło tam siedzieć? Wprawdzie mamy na wyposażeniu ICP-OES, ale włączanie aparatu na jedną próbkę było bezsensowne, najwyżej dołożymy próbkę roztworzonego osadu do innych, gdy będzie więcej zamówień. Na szybko to ja mogłem zrobić tylko klasyczne analizy próbówkowe.
Przegrzebałem odczynniki, szukając czegoś specyficznego. Dimetyloglioksymu nie było, podobnie jak benzoinooksymu, rodanek amonu jeszcze nie doszedł, wreszcie zdecydowałem się na dwie reakcje. Do części roztworu dodałem wodorotlenku sodu - powstał jasnoniebieski osad, jaki raczej daje miedź. Do drugiej dodałem roztwór żelazocyjanku potasu (ściślej: odczynnik Carreza I), powstał brunatno-czerwony osad, charakterystyczny dla miedzi. Aha, czyli w sumie potwierdzone.
Zastanawiałem się jednak czy da się w tym jeszcze wykryć żelazo, które zdecydowanie częściej występuje w wodzie, podbarwiając kamień kotłowy na brązowo. Nie było widać aby osad wodorotlenku podbarwiał rdzawy wodorotlenek żelaza III. Obstawiałem, że żelazo może być ewentualnie związane w formie związków żelaza II o słabym, zielonkawym zabarwieniu, maskowanym przez miedź. Postanowiłem dodać do zlewki kilka kropel wody utlenionej. Żelazo się dzięki temu utleni, osad pociemnieje. Myślałem przy tym, że wodorotlenek miedzi nie zareaguje, bo aby utlenić miedź do jeszcze wyższych stopni utlenienia, to trzeba bardziej agresywnych warunków. A jednak zostałem zaskoczony - zawartość zlewki stała się intensywnie zielona:
No dobra, może to żelazo daje taki efekt. Żółty i niebieski dają zielony, widocznie żelaza jest w tym osadzie dużo. Zrobiłem ślepą próbę z siarczanem miedzi, ale wynik był podobny - wodorotlenek miedzi w reakcji z wodą utlenioną robił się zielony.
Wyjaśnienie wiąże się z powstawaniem związku, o jakim wcześniej nie czytałem - powstaje nadtlenek miedzi - CuO2. Jest to związek nietrwały, szybko zresztą zaobserwowałem stopniowe ciemnienie i brunatnienie osadu, zachodzące z wydzielaniem bąbelków tlenu; po upływie 30 minut w zlewce był już tylko drobny, czarny osad tlenku miedzi II CuO.
Charakter i struktura tego tlenku są dyskutowane - w pewnym warunkach daje się uzyskać związek o podobnej stechiometrii, mający charakter dwutlenku, z czterowartościową miedzią.[1] W tym przypadku przypuszczalnie jest to cykliczny nadtlenek jonowy, zawierający dwuanion O2− 2 .
W strukturze wewnętrznej wysokotemperaturowych nadprzewodników z grupy miedzianów, daje się wyróżnić płaszczyzny tlenku o stosunku 1:2, w których następuje nadprzewodnictwo elektronowe.
Tymczasem następnego dnia zauważyłem, że w próbówce w której badałem reakcję na żelazocyjanki, roztwór nad osadem związków miedzi zabarwił się na niebiesko. Błękit pruski jest lepiej rozpuszczalny w wodzie od żelazocyjanku miedzi, zwłaszcza przy mocno niestechiometrycznym stosunku jonów, co świadczyło o tym, że kamień kotłowy jednak zawierał też trochę żelaza.
ps. Tak się złożyło, że to 300 wpis na tym blogu.
--------
[1] https://pubs.acs.org/doi/10.1021/j150655a014
Widoczek przypomina trochę kolonie pleśni na odżywce, ale ma zupełnie inną naturę. O tym ciekawym fenomenie dowiedziałem się niedawno, szukając informacji na temat innej reakcji. Publikacja sprzed 10 lat* opisywała nowy rodzaj samoorganizacji w procesach chemicznych. Gdy kryształ soli zostaje umieszczony w cienkiej warstwie roztworu, w którym wytrąca osad, w niektórych przypadkach osad nie tworzy po prostu równego kółeczka, sięgającego do miejsca, w którym stężenie powstających związków jeszcze wystarczało na wytrącenie. Zamiast tego powstają promienie, w których strąca się więcej osadu, przedzielone obszarami bez wytrąceń. Dokładny wzór zależy od warunków.
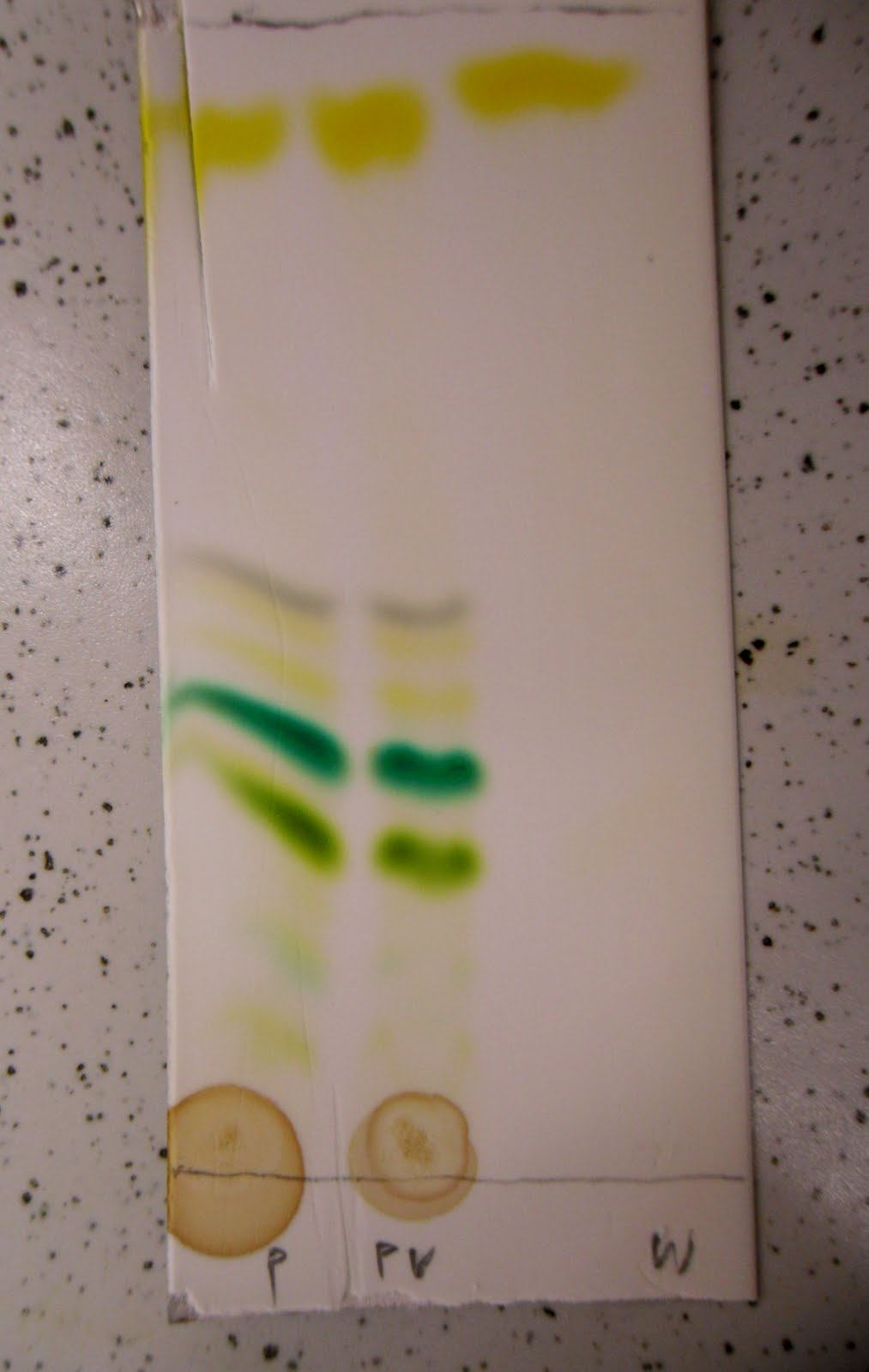
Efekt zauważono w przypadku soli miedzi i kobaltu w warstwie roztworu szczawianu sodu. Takie też zestawy testowałem. W powyższym przypadku sole miedzi (na dole) dały bardzo słaby efekt, w dodatku wzór rozmył się po przeniesieniu szalki na ciemniejsze tło. Natomiast kobalt dał wyraźny wzór rzadko rozłożonych promieni strzelających z okrągłej plamki osadów.
Przy kolejnym powtórzeniu, z innym stężeniem szczawianu i z cieńszą warstwą roztworu, wzór dla kobaltu wyszedł dużo gęstszy i mniej czysty:
W innej porcji tego samego roztworu sól miedzi także dała gęsty wzór:
Po pewnym czasie osady były na tyle dobrze związane z podłożem, że można było wyssać roztwór pipetką.
Skąd ten efekt? Autorzy artykułu podają, że to wynik nałożenia się kilku zjawisk. Rozchodzenie się jonów wokół kryształka nie następuje tylko na drodze prostej dyfuzji. Początkowo tworzy się znacznie gęstszy roztwór danej soli, który rozprzestrzenia się tuż przy dnie. Generuje to w warstwie roztworów ruch warstw - przydennej na zewnątrz a powierzchniowej do środka. Pierwsze porcje wytrącającego się osadu wprowadzają w ten ruch zaburzenia. Ostatecznie zawirowania porządkują się w układ podobnej wielkości poziomych wirów, rozciąganych ruchem rozpływającej się warstwy przy dnie. W tak ukształtowanych strumieniach wytrąca się więcej osadu.
Był to efekt nieoczekiwany, zamierzeniem autorów było badanie tworzenia się osadu w przepływie grawitacyjnym, powstawanie kolistych wytrąceń wokół miejsca rozprzestrzeniania roztworu wytrącanego było znane, tymczasem po uzyskaniu odpowiednich warunków stężeń i grubości roztworu powstawały promienie, których dotychczas w literaturze nie opisywano.
W obrębie jednolitego koła kryształki szczawianów miały normalną formę wielościennych kryształów i ich kulistych skupień. W promieniach natomiast tworzyły włókna o grubości 3 mikrometrów i długości do jednego czy nawet kilku milimetrów. Dosłownie włos z pojedynczych kryształów.
Badacze wykazali pojawianie się tego efektu w dość rozcieńczonych roztworach mrówczanu (0,01-0,03M) , w za bardzo stężonych cała rozpuszczana sól formowała jednolite koło albo osad oblepiał wrzucony kryształek. Mam więc w sumie szczęście, że udało mi się ten efekt odtworzyć, bo stężenia były dobierane na oko.
----------
* J. Maselko et al., Precipitation Pattern Formation in the Copper(II) Oxalate System with Gravity Flow and Axial Symmetry, J. Phys. Chem. A, 2009, 113 (29), pp 8243–8248
Jak robić roztwór jodu, gdy nie ma jodu? Na taki problem natknąłem się ostatnio podczas oznaczania węglowodanów ogólnych w pewnej próbce. Typową metodą, w sytuacji gdy nie zależy nam na oznaczeniu błonnika (lub go tam nie ma) jest hydroliza skrobi i dekstryn do glukozy i oznaczenie cukrów redukujących którąś z metod miareczkowych. Aby zhydrolizować skrobię w próbce, trzeba ją odpowiednio długo gotować z kwasem solnym, sprawdzając co pewien czas roztworem jodu, czy skrobia już przereagowała.
Jednak po przejrzeniu wszystkich szafek wyszło na to, że ani jodyny ani płynu Lugola nie mieliśmy na stanie. Co tu robić? A no zsyntezować jod i go sobie rozpuścić. Dokonałem tego przy pomocy metody, która nie jest nowością, ale ją sobie kiedyś na pracowni magisterskiej w takiej procedurze wymyśliłem. Całość opiera się na tej samej reakcji, co przy jodometrycznym oznaczaniu miedzi - sole miedzi II reagują z jonami jodkowymi z wytworzeniem jodku miedzi II, który szybko dysproporcjonuje do wolnego jodu i jodku miedzi I. Wynika to stąd, że jony miedzi II są dość dobrym utleniaczem, zaś jony jodkowe chętnie się utleniają do wolnego jodu:
Cu2+ + 2I− → CuI2
2 CuI2 → 2 CuI↓ + I2
Procedura którą zastosowałem polegała na przeprowadzeniu tej reakcji w zasadzie na sucho. Reakcja w takiej formie jest na tyle efektowna, że mogłaby być elementem doświadczeń pokazowych.
Tak więc, do porcelanowego moździerza wsypujemy niewielkie ilości jodku potasu i siarczanu miedzi:
I zaczynamy je ucierać. Zmiana koloru na brązowy następuje natychmiast po zmieszaniu roztartych proszków:
Po dokładnym roztarciu masa zamienia się w gęstą ciecz, zawierającą stały osad, tworząc czekoladowe błotko o ostrym zapachu:
które można już teraz rozpuścić. Ja użyłem alkoholu, jeśli do reakcji użyje się wyraźnego nadmiaru jodku potasu, będzie można użyć wody.
Jest to prosty przykład reakcji w fazie stałej, w której substancje reagują ze sobą podczas ucierania. Proces sam przyspiesza w związku z uwolnieniem z siarczanu miedzi wody krystalizacyjnej (5 moli na jeden mol miedzi), która rozpuszcza reagenty, ułatwiając im przereagowanie do końca. Po zdekantowaniu roztworu jodu pozostaje nam kremowy osad jodku miedzi I, który ma swoją drogą ciekawe zastosowania w chemii organicznej, jako środek jodujący.
Po podgrzaniu i odparowaniu powstałej wody, wydzielony jod można oczyścić przez resublimację.
Całkiem na sucho, czyli z bezwodnym siarczanem miedzi, reakcja powinna zachodzić wolno, jeśli w ogóle.
Kolejny przypadek pojawienia się czegoś ciekawego w kolbie obok właściwej syntezy.
W zeszłym tygodniu obrabiałem mieszaninę po reakcji, w której użytym katalizatorem były sole miedzi. Aby usunąć z mieszaniny ją i kwaśne produkty uboczne, mieszaninę rozpuszczoną w octanie etylu ekstrahowałem roztworem węglanu sodu a potem solanką dla odciągnięcia wody. Miedź ładnie przechodziła do fazy wodnej:
Roztwór węglanu, brany z butli, w której uprzednio go przygotowałem, był niemal stężony, solanka podobnie, a ponieważ z fazy organicznej przeszedł jeszcze rozpuszczalny w wodzie acetonitryl, szybko w kolbie ze zlewkami wytrącił się osad węglanu, nad którym pozostał granatowy roztwór soli miedzi:
Kolbkę odstawiłem w kąt dygestorium i nie zajmowałem się dalej. Po weekendzie okazało się, że trochę roztworu odparowało, a z mieszaniny zaczął krystalizować osad w formie drobnych igiełek
Najwięcej zebrało się na dnie, na białej warstwie węglanu. Było to dość ciekawe. Roztwór był zasadowy, zawierał jony miedziowe, węglanowe, hydroksylowe i chlorkowe, i był właściwie nasycony. W tych warunkach wypaść z niego mogło kilka niebieskich substancji - węglan miedzi, chlorek miedzi, zasadowy węglan miedzi, zasadowy chlorek miedzi, wodorotlenek miedzi...
Aby coś rozstrzygnąć (i pobawić się) ostrożnie zlałem roztwór, zostawiając tylko zmieszane osady:
Przepłukałem je następnie zimną wodą, aby wypłukać dobrze rozpuszczalny węglan sodu. Sam niebieski osad był w wodzie raczej słabo rozpuszczalny, co raczej wykluczało chlorek miedzi. Po odsączeniu małą ilość osadu przeniosłem na szkiełko zegarkowe i zadałem paroma kroplami rozcieńczonego kwasu solnego. Osad rozpuścił się, do ostatniej grudki wydzielając bąbelki gazu. Zatem musiał to być jakiś węglan miedzi. A konkretnie azuryt.
Miedź na drugim stopniu utlenienia tworzy z jonami węglanowymi sole, jednak w normalnych warunkach nie jest to po prostu stechiometryczny węglan CuCO 3 , jak by się to mogło wydawać (i jak jest to zwykle zapisywane w podręcznikach). Wynika to z większego powinowactwa jonów hydroksylowych niż węglanowych, które słabo koordynują. W praktyce więc z wodnych roztworów wytrąca się zasadowy węglan miedzi, zawierający jony węglanowe i wodorotlenkowe. Ten jednak może występować w dwóch formach, różniących się ilością jonów węglanowych. Forma zawierająca dwa jony węglanowe i dwa hydroksylowe (Cu3(CO3)2(OH)2 ) to błękitny azuryt, zaś forma z jednym jonem węglanowym i dwoma hydroksylowymi ( Cu2CO3(OH)2 ) to zielony malachit. Ich trwałość zależy od warunków - pod ciśnieniem atmosferycznym trwalszy jest malachit, zawierający mniej jonów węglanowych. Azuryt wymaga większego ciśnienia dwutlenku węgla dla uzyskania równowagi, bez tego, zwłaszcza pod wpływem wilgoci, ulega powolnej przemianie w malachit. Często więc w złożach obserwuje się oba minerały zmieszane, z ziarnami azurytu zachowanymi wewnątrz brył malachitowych.
Podczas wytrącania węglanu miedzi w trakcie szkolnych eksperymentów zwykle otrzymuje się osad zawierający przewagę malachitu, o niebiesko-zielonym kolorze, wynika to z niedostatecznej ilości jonów węglanowych. Dalsze przetrzymywanie mokrego osadu wywołuje po pewnym czasie całkowitą konwersję w malachit, przeprowadzenie reakcji w podwyższonej temperaturze przyspiesza przemianę do jednej-dwóch godzin.
Przypuszczam, że w tym przypadkowym osadzie otrzymałem raczej przewagę azurytu. Sprzyja temu przeprowadzenie krystalizacji w nasyconym roztworze węglanu (duży nadmiar jonów węglanowych). Jedną ze wskazówek jest wyraźnie niebieski kolor osuszonego osadu:
Azuryt występuje w naturze jako minerał wtórny towarzyszący rudom miedzi, zwykle wraz z malachitem. Już od starożytności budził zainteresowanie, ze skał z przewagą tego składnika wykonywano ozdoby, zaś on sam po rozkruszeniu służył jako pigment malarski. Niestety znaną niedogodnością było powolne zielenienie oraz mała odporność na kwaśne składniki farby lub spoiwa, dlatego bardziej ceniona była ultramaryna, otrzymywana z minerału lapis lauzuli.
Malachit syntetyczny, otrzymywany z roztworów, znany był jako verditer, a syntetyczny azuryt jako niebieski verditer.
Neutralny węglan miedzi, jaki wydawałoby się, że powinien powstawać w reakcji, jest natomiast związkiem nietrwałym w obecności wilgoci i trudno otrzymać go normalną drogą. W zasadzie dopiero w 1973 roku opublikowano syntezę w bezwodnych warunkach między zasadowym węglanem a dwutlenkiem węgla, w wysokiej temperaturze i ciśnieniu. Ma postać szarego proszku i nie znalazł jakiegoś specjalnego zastosowania. [a]
--------
[a] Hartmut Erhardt, Wilhelm Johannes, and
Hinrich Seidel (1973): "Hochdrucksynthese von Kupfer(II)-Carbonat", Z.
Naturforsch., volume 28b, issue 9-10, page 682.
Przygotowałem roztwór badanej substancji w deuterowanym DMSO, przeniosłem do czystej szklanej fiolki, opisałem na papierowej etykietce jakie jest jej oznaczenie i zaniosłem do pomieszczenia ze sprzętem do NMR. I na miejscu stwierdziłem, że próbka znowu mi zamarzła.
Gdy pierwszy raz mi się to trafiło, sądziłem że badana substancja wykrystalizowała. Nie jest to dobra sytuacja. Do badania bierzemy nieco mniej niż mililitr roztworu, który musi zawierać kilka miligramów badanej substancji, ze względu na czułość aparatu. Substancja nie zawsze łatwo się rozpuszcza, dlatego czasem faktycznie zdarza się, że zaczyna ponownie krystalizować w próbówce. Takie próbki dają widma mniej przydatne, z poszerzonymi sygnałami grup, czasem z dodatkowymi rozszczepieniami, co utrudnia interpretację.
W tym przypadku na szczęście było inaczej, na korytarzu było dosyć chłodno i rozpuszczalnik zamarzł. Temperatura krzepnięcia dimetylosulfotlenku to 18 °C, deuterowanego nawet więcej bo 20 °C. Wystarczy więc temperatura nieco niższa od pokojowej aby zaczął zamarzać. Wystarczyło ogrzać próbkę w dłoni. Kolejka na urządzeniu była krótka, a próbka wewnątrz samplera nie wychłodziła się na tyle aby zakrzepnąć ponownie.
Ostatnio w laboratorium przypadkiem uzyskałem efekt, który wcześniej trudno mi było uzyskać zamierzenie w domowych warunkach. Podczas oczyszczania mieszaniny poreakcyjnej stosowałem między innymi ekstrakcję z roztworem wodorowęglanu sodu, dla usunięcia kwaśnych produktów ubocznych, po czym dla zmniejszenia zawartości wody w fazie organicznej przeekstrahowałem ją jeszcze z nasyconą solanką. Nasycony roztwór soli chętnie chłonie wodę. Oczywiście do ostatecznego dosuszenia używa się desykatorów w rodzaju bezwodnego siarczanu sodu, ale bez wstępnego odciągnięcia nadmiaru wody solanką, należałoby użyć tego środka wiążącego całkiem sporo.
Tak więc po rozdzieleniu faz oddzieliłem organiczną od solanki, którą to zlałem do osobnej zlewki. Tą odłożyłem na bok, i zapomniawszy wylać zostawiłem na weekend. W poniedziałek stwierdziłem, że mimo iż na ściankach nie było widać śladów krystalizacji czy większego odparowania, to na dnie zlewki powstało kilka ładnie uformowanych kryształów:
Choć sól kuchenna jest jednym z pierwszych wyborów, gdy tylko przyjdzie nam do głowy robienie sobie w domu kryształków, niespecjalnie się do tego nadaje. Ma niestety skłonność do krystalizowania w zbitych skupiskach drobnokrystalicznych, a różnica rozpuszczalności w wodzie między temperaturą pokojową a wrzątkiem jest mała, przez co wymuszanie krystalizacji przez przesycenie spadkiem temperatury, jest mało efektywne. W większości przypadków próby krystalizacji polegające na zanurzeniu sznurka w solance, kończą się powstaniem czegoś podobnego do białego stalaktytu.
Podczas domowych prób rezygnowałem więc z krystalizacji na sznurku czy nitce, zamiast tego wrzucałem do roztworu jakiś obiekt, który pozostawał całkowicie zanurzony.
Ziarno czarnego pieprzu z przezroczystymi kryształami
Teraz powstało mi natomiast bez żadnych przygotowań, bez powstania drobnokrystalicznych osadów i innych przeszkadzających dodatków, kilka dość regularnych kryształów o wielkości do pół centymetra:
Oczywiście, jak widać, nie są idealne. Są białe, zapewne podczas szybkiego wzrostu pochłaniały z roztworu zanieczyszczenia organiczne i banieczki powietrza. Rosły przyrośnięte do dna, dlatego zamiast regularnych kostek powstały płytki. Ciekawą cechą jest też kształt górnej ściany krystalicznej - posiada regularne wgłębienie, zagłębiające się w kryształ równymi schodkami.
To tak zwany kryształ szkieletowy, powstający w warunkach szybkiego wzrostu w warunkach przesycenia. Woda odparowywała z solanki, bardziej zagęszczony roztwór opadał na dno. Kryształy powstawały więc w warstwie nieruchomego roztworu o dużym stopniu przesycenia. Grawitacja w przypadku górnej ścianki kryształu nie wspomagała ruchu cieczy, zatem szybkość krystalizacji była ograniczana szybkością dyfuzji cząstek, oraz różnicowana anizotropią budowy kryształu.
W takich sytuacjach częstym zjawiskiem jest szybszy przyrost kryształu na krawędziach niż pośrodku ściany, wynika to z lepszej dostępności roztworu (zamiast docierać z połowy przestrzeni, cząstki mają 3/4 objętości z której dochodzą do powierzchni kryształu). Efekt ten może wzmacniać pojawiające się na krysztale pole elektryczne.
Zależnie od tego jak duży jest stopień przesycenia, oraz jaki jest normalny pokrój kryształów danej substancji, tworzą się więc albo kryształy zachowujące normalny pokrój, ale ze schodkowatymi lejami na ścianach, albo mające formę cienkich lejków, albo przybierające kształty igieł, gwiazdek czy pierzastych dendrytów. Z tworzenia takiego pokroju znany jest właśnie halit, ale obserwowano go też u kwarcu, kalcytu i w kryształach lodu. Łatwo powstaje też podczas krystalizacji bizmutu, metalu o stosunkowo niskiej temperaturze topnienia, z którego da się w amatorskich warunkach uzyskać efektowne kryształy.
Wraz ze zmianą warunków, forma kryształu może się zmienić. Zmniejszenie stopnia przesycenia roztworu często powoduje wypełnienie jam w ścianach, jedyną pozostałością mogą wówczas być wtrącenia układające się wewnątrz kryształu w stożkowaty wzór.
Gdy chemik wychodzi z pracy do domu, dobrze jest zadbać o to, aby przypadkiem nie wynosił na sobie chemikaliów. Zwłaszcza na rękach, którymi będzie potem dotykał wszystkiego, w tym domowników i siebie. Oczywiście przy pracy z odczynnikami powinno się stosować rękawiczki, ale trudno się zupełnie ustrzec przed pobrudzeniem. Często przed wyjściem do domu, po umyciu rąk sprawdzam ich czystość pod lampą ultrafioletową ustawioną na dalszy, "czarny" zakres. Większość substancji z którymi pracuję, w jakimś stopniu świeci w takim zakresie, zwykle na niebiesko lub żółto.
Dlatego łatwo będzie zrozumieć moje obawy, gdy podczas takich prób stwierdziłem wyraźną fluorescencję samych paznokci, zupełnie jakby czymś się nasączyły:
Dopiero przegląd literatury nieco mnie uspokoił. Otóż wygląda na to, że paznokcie fluoryzują same z siebie. W przeglądzie dermatologicznych badań nad skórą opisano naturalne świecenie naświetlanych ultrafioletem paznokci.[1] Nie dowiedziałem się natomiast co konkretnie w nich świeci, może bilirubina która dość często w stanach chorobowych odkłada się w płytce paznokcia aż do wyraźnego zabarwienia.
Do paznokci mogą też przenikać fluoryzujące leki - po zażyciu tetracykliny obserwuje się żółte świecenie, a po zażyciu atabryny żółto-zielone.[2] Próbuje się wykorzystać to zjawisko w bezinwazyjnej diagnostyce.
---------
[1] Pierre Agache, Philippe Humbert, Measuring the Skin, Google Books s. 296
W tym przypadku była to pewna pochodna chinoliny. Kilkakrotna krystalizacja, rozpuszczenie i ponowna krystalizacja pozwoliły na oddzielenie brązowego zanieczyszczenia od jasnokremowego związku bez konieczności użycia chromatografii.
Kiedyś już pisałem o jednej z podstawowych technik oczyszczania mieszanin poreakcyjnych - preparatywnej chromatografii kolumnowej, przy pomocy własnoręcznie napełnianej żelem krzemionkowym szklanej kolumny. Teraz natomiast w firmie mam okazję częściej popracować na sprzęcie, z jakim jako prosty magistrant nie miałem bezpośrednio kontaktu - z automatem do szybkiej ("flashowej") chromatografii kolumnowej.
Jest to coś pośredniego między klasyczną chromatografią na szklanych kolumnach a wysokosprawną HPLC, służy do rozdziałów mieszanin na skalę preparatywną. Używa się do niej najczęściej gotowych kolumn, nabitych dobrze upakowanym, suchym żelem krzemionkowym. Po nałożeniu mieszaniny na początek kolumny, najlepiej przez nastrzyk roztworu, podłącza się ją do automatu, który pod umiarkowanym ciśnieniem przepuszcza przez nią eluent o zaprogramowanym składzie. Możliwe jest ustalenie programu elucji, z określonymi gradientami zmian składu roztworu w czasie procesu, ewentualnie też zmianami szybkości przepływu.
Aparat wykrywa detektorem UV-VIS pojawienie się frakcji w odcieku i po przekroczeniu zadanego progu zbiera frakcje do próbówek zgodnie z określoną kolejnością.
Oczywiście dalszą robotą nad ustaleniem, która frakcja jest substancją nam potrzebną musi się już zająć laborant, ale sam proces nie wymaga ciągłego nadzoru.
Dostępne są kolumny różnych wielkości, dobierane odpowiednio do ilości rozdzielanych mieszanin, największa na jakiej na razie robiłem miała objętość 300 ml i posłużyła do rozdziału porcji 7 g, ale widziałem w katalogu nawet kolumny ponadlitrowe.
Gdy zakończyłeś przeprowadzać reakcję w dużej skali i rankiem następnego dnia od razu wiesz, że produktu będzie dużo:
Seria z obrazkami z labu wraca, bo znów zacząłem pracować w dziedzinie. Po kilku miesiącach przestoju po rezygnacji z doktoratu na UW dostałem się do firmy Selvita S.A. a konkretnie do niedawno otworzonego oddziału w Poznaniu. Firma zajmuje się głównie syntezami farmaceutyków na zamówienie oraz wstępnymi badaniami (in silica/in vitro), inną gałęzią jest szukanie kandydatów na leki na własny rachunek, dwie perspektywiczne substancje trafiły już do testów klinicznych.
Moje stanowisko polega na syntezach organicznych na skalę nieco większą niż to miałem do czynienia na uczelniach.
Oddział w Poznaniu jest jeszcze mały, nowi pracownicy są nadal poszukiwani, na razie panuje tam raczej towarzyska atmosfera (o oddziale w Krakowie czytałem raczej odmienne opinie). Siedziba laboratoriów mieści się na terenie kampusu Morasko UM, zaraz obok wydziału chemii, więc jeśli czytają mnie studenci stamtąd, to pozdrawiam.
Zdjęcie przedstawia wygląd mieszadła w reaktorze po przeprowadzeniu sprzęgania Suzuki. Jest to reakcja tworzenia wiązania węgiel-węgiel pozwalająca skleić nową cząsteczkę z różnych kawałków. Jednym z substratów są związki boroorganiczne a drugim halogenki, reakcję katalizuje pallad oraz słabe zasady. W moim przypadku łączone części były dość rozbudowanymi związkami aromatycznymi, wytwarzane było bezpośrednie wiązanie między pierścieniami, reakcja zaszła niemal ilościowo a produkt można było wydzielić przez krystalizację z mieszaniny poreakcyjnej. Po wyłączeniu mieszania i grzania, rankiem kolejnego dnia zastałem widok jak na obrazku.
Publikowałem tu już kiedyś zdjęcia widm lamp jarzeniowych z gazami szlachetnymi, teraz wrzucę jeszcze jedno z tamtych ćwiczeń, pokazujące linie emisyjne wodoru w paśmie widzialnym:
Wyraźnie widoczne są trzy pasma, tzw. "seria Balmera": czerwone o długości fali 656 nm, zielonkawo-niebieskie 486 nm i niebieskofioletowe 434 nm. Nieco bardziej na lewo powinno być jeszcze czwarte pasmo i nawet gołym okiem było je słabo widać, ale na aparacie przy tej jasności lampy się nie załapało, ma długość fali 410 nm i leży na pograniczu widzialnego fioletu i ultrafioletu. Poza zasięgiem aparatu pojawiają się jeszcze pasma ultrafioletowe.
Aparat cyfrowy nie do końca dobrze oddał kolory, środkowe pasmo było patrząc gołym okiem nieco bardziej morskie, dodatkowe widmo w tle miało szerszy zakres żółty a czerwone pasmo nie było tak mocno jasne, aż prześwietlone.
Uzupełniające tło słabsze widmo wielu drobnych linii to prawdopodobnie efekt domieszki azotu w wodorze wypełniającym rurkę jarzeniową.
Linie emisyjne wodoru odegrały dużą rolę w chemii kwantowej. Balmer znalazł ogólny wzór opisujący długość fali poszczególnych linii za pomocą pewnego prostego szeregu. Była to w zasadzie czysta numerologia, czyli dobieranie liczb tak, aby pojawił się między nimi związek, ale rozszerzenie tej formuły pozwoliło przewidzieć odkryte później linie w zakresie ultrafioletu. Wzór został uogólniony przez Rydberga i okazał się przewidywać także linie emisji w dalekim ultrafiolecie, dalszej podczerwieni.
Gdy zaczęła się formować teoria atomu jako układu złożonego z dodatnio naładowanego jądra i elektronów krążących wokół, serie widmowe znalazły wyjaśnienie jako emisje w postaci światła energii, będącej różnicą między energią kolejnych coraz bardziej oddalonych orbit, wydzielaną przez elektrony wzbudzone, spadające z dalekich orbit na te bliższe jądru. Bardzo wąskie linie emisji oznaczają, że zakresy energetyczne dla orbit są dość ściśle określone, zostało to dobrze wyjaśnione dopiero na gruncie kwantowego modelu atomu (w modelu Bohra dozwolone orbity były stacjonarne, bo tak).
Widmo wzbudzonego wodoru było przy tym o tyle łatwe do opisu, że wynikało z najprostszego możliwego układu z jednym elektronem. Opis widm innych pierwiastków nie jest tak łatwy i tylko do części linii widmowych stosuje się wprost wzór Rydberga.
ps. a nowe materiały z laboratorium nie pojawiają się, bo już nie pracuję na UW.
Wielokrotnie w różnych wpisach pokazywałem wygodną i szybką metodę sprawdzania składu mieszanin poreakcyjnych, czyli chromatografią cienkowarstwową na wycinanej z arkusza płytce:
Jest prosta, pozwala dobrać skład eluentów, oraz często oddzielone składniki są bardzo ostro widoczne. Niemniej powtórzenie tego samego procesu z identycznym eluentem na kolumnie, często nie daje tak ładnych rezultatów. Oddzielone porcje podróżując wzdłuż kolumny rozmywają się i czasem zaczynają wtórnie na siebie zachodzić. Rozdział nie jest więc tak dobry jak to wyglądało na płytce.
Jednym z pomysłów na to jak rozwiązać ten problem, jest wykonanie rozdziału na bardzo dużej płytce - w systemie TLC preparatywnej:
Rozdzielony wyciąg z liści, widoczne pasma chlorofilu, karotenoidów i fityn
Taka płytka ma formę szklanej tafli o boku kilku lub kilkunastu centymetrów z nałożoną dość grubą warstwą podłoża rozdzielającego. Przy pomocy kapilarki lub pipetki nad dolną krawędzią płytki nakłada się poziomą krechę mieszaniny rozdzielanej, wielokrotnie powtarzają nakładanie. Następnie tak samo jak w małych płytkach, dolną krawędź zanurza się w eluencie. Potrzebna jest do tego odpowiednio duża komora, ja w jednym takim przypadku użyłem komory wielkości małego akwarium. Gdy płytka nasiąknie, krecha mieszaniny rozdziela się na długie pasy, zawierające oddzielone składniki. Aby je teraz ostatecznie oddzielić, bierze się nożyk lub szpatułkę o ostrym brzegu, i wydłubuje ten składnik, o jaki nam chodzi, zdrapując go ze szkła wraz z podłożem:
Zdrapiny zalewa się następnie jakimś mocnym eluentem aby wymyć oddzieloną frakcję. Można w ten sposób rozdzielać do około 0,5-1 g mieszaniny poreakcyjnej.
Ponoć można zdrapywać też plamki ze zwykłych, małych płytek, do celu badań jakąś bardzo czułą metodą analityczną, gdy dysponujemy małą ilością mieszaniny, wtedy do rozdziału wystarcza jedna kropla. Sam nigdy tego nie robiłem, ale słyszałem, że niektórzy się tak bawią.
Jednym z obowiązków doktorantów jest przeprowadzenie odpowiedniej ilości godzin dydaktycznych ze studentami. W zeszłym roku pomagałem przy preparatyce organicznej, w tym natomiast przy zajęciach z fizyki.
Jedną z zalet tych zajęć było to, że mogłem jeszcze raz samemu przyswoić sobie pewne podstawy. Oraz że czasem miałem okazję zrobić ładne zdjęcia. Tak było podczas prowadzenia ćwiczenia ze spektroskopii - student na stole mierzył spektroskopem kąty ugięcia poszczególnych prążków emisyjnych emitowanych przez lampy z różnymi gazami, a ja próbowałem jakoś ładnie to uchwycić:
Najlepiej wyglądało to przy użyciu siatki dyfrakcyjnej lustrzanej, dającej jasne obrazy. Tutaj lampa ze świecącym helem:
Ostatnio w ramach specjalizacji z krystalografii próbowałem wykrystalizować i wstępnie zbadać kokryształ glicyna+kwas glutarowy. Akurat z samą krystalizacją nie było zbyt dużego problemu:
Kokryształy to kryształy tworzone przez równocześnie dwie lub więcej substancji tworzących powtarzalny układ w sieci. Zwykle precyzuje się, że różne cząsteczki oddziałują niejonowo, co odróżnia je od soli, oraz że obie substancje tworzą czyste kryształy w warunkach istnienia kokryształu, co odróżnia je od hydratów i części klatratów.
Dla danego typu połączenia substancje składowe zachowują stały stosunek stechiometryczny, na przykład dla kryształu chinhydronu na jedną cząsteczkę hydrochinonu przypada jedna cząsteczka chinonu, z którym tworzy kompleks.
Zachodzenie kokrystalizacji może wynikać bądź z tworzenia nowej struktury, bądź z możliwości wpasowania się jednej, podobnej rozmiarami cząsteczki, w normalną sieć krystaliczną drugiego związku. Obecnie temat jest intensywnie badany na potrzeby farmaceutyki, bowiem kokryształy leków mogą być bardziej trwałe, trudniej topliwe lub wykazywać odmienną rozpuszczalność niż substancja czynna w formie czystej.[1]
W tym przypadku sposób otrzymania był prosty, lecz nie do końca pewny - rozpuściliśmy po prostu w wodzie glicynę i kwas glutarowy w stosunku molowym 1:1, nieco ogrzaliśmy dla odparowania aby otrzymać roztwór przesycony i wylaliśmy niewielką ilość na szklaną szalkę. Składniki wykrystalizowały w formie igiełkowatych kryształków i pozostawało tylko dobrać do badania odpowiedni, oraz trafić na kokryształ, bo kształtem nie wyróżniał się od kryształów glicyny i kwasu glutarowego które także mogły powstać.
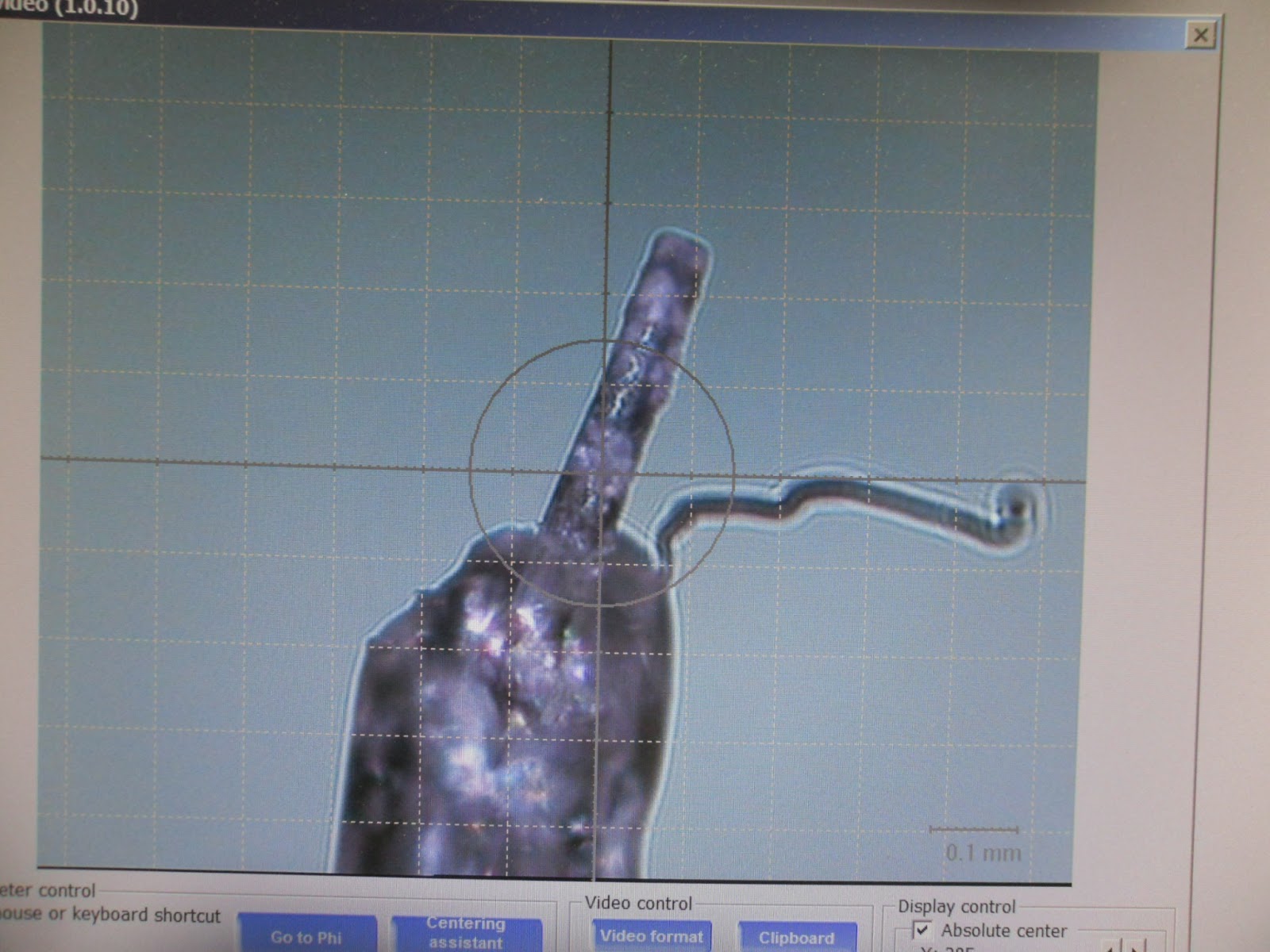
Układ pomiarowy w dyfraktometrze: na końcu szklanej kapilary mały jasny punkcik to kryształ, z lewej strzela wiązka promieniowania, powyżej kamera pozwalająca wypośrodkować kryształ i oświetlenie. Po prawej obudowa detektora.
Pierwszy zbadany kryształek prawdopodobnie należał do poszukiwanego połączenia ale nie dawał sygnału odpowiedniej jakości aby to potwierdzić - był trochę za duży i oprószony drobniejszymi na powierzchni. Ze względu na rozmiar wiązki promieni rentgenowskich, kryształ powinien być mniejszy niż milimetr, oraz powinien być monokryształem bez spękań i przyrośniętych bliźniaków.
Należało więc poszukać następnego. Odpowiednio wyglądającą igłę przyciąłem manipulując pod mikroskopem pęsetą i skalpelem, aby nie była zbyt duża, i przykleiłem na kropelkę do kapilary na końcu główki goniometru. Tym razem szybkie sprawdzenie pokazało sygnał dość dobrej jakości, ale inny niż oczekiwany. Wyselekcjonowany kryształek okazał się glicyną w odmianie alfa.
Zostało mało czasu, więc wybrałem z szalki mały kryształek, którego nie trzeba było ciąć. Niestety podczas próby nałożenia złamała się szklana kapilara do której przyklejane były kryształy. Ostatecznie postanowiliśmy zaryzykować i kryształek został przyklejony do zachowanego kikuta, trochę grubego, ale jeszcze dostatecznego. Kryształ przykleił się krzywo a w dodatku obok przyczepił się czyjś włos. Nie wyglądało to dobrze:
Jednak wstępny pomiar pokazał, że kryształ jest bardzo dobrej jakości, bez zakłóceń, i ma inną grupę przestrzenną. Dzięki temu mogliśmy sprawdzić parametry komórki krystalicznej w bazie i stwierdzić że jest to... glicyna w formie beta.
Odmiana beta jest metastabilna w normalnych warunkach i zwykle szybko zamienia się w formę alfa. Na zajęciach ze studentami kilka miesięcy wcześniej żadnej grupie nie udało się jej uzyskać, a teraz jak na złość. Ponieważ kończył się czas, dalszą krystalizację odłożyliśmy do następnych zajęć.
----------
[1] http://biuletynfarmacji.wum.edu.pl/1305Sokal/Sokal.html
Kryształy białka lizozymu, otrzymane metodą wiszącej kropli:
Lizozym to enzym bakteriobójczy, powodujący rozkład glikoprotein w ścianie komórkowej bakterii. Po uszkodzeniu ściany następuje pęknięcie i rozpłynięcie się komórki bakterii, czyli tzw. liza, stąd nazwa enzymu. Jego cząsteczka składa się ze 129 aminokwasów.
Lizozym należy do białek które stosunkowo łatwo krystalizują, dlatego wcześnie poznano jego strukturę.
Kryształy na zdjęciach są takie sobie, przede wszystkim bardzo małe, może za parę dni bardziej urosną. Natomiast zachwyciła mnie gra kolorów widoczna pod filtrem polaryzacyjnym. Jestem mimo wszystko trochę estetą.
* * *
Myślę, że dobrze by było przy tej okazji parę rzeczy objaśnić, bo może regularnie zaglądający zastanawiali się nad paroma kwestiami. Ponieważ prace typowo syntetyczne na pracowni prof. Czarnockiego nie bardzo mi wychodziły, ostatecznie zmieniłem promotora i temat pracy, bo istniało ryzyko, że ciągnąc dotychczasowy mógłbym nie otrzymać wyników na satysfakcjonującą pracę doktorską.
Tak że teraz zająłem się czymś bliższym chemii fizycznej, czyli badaniami krystalograficznymi. Prawdopodobnie ostateczny temat będzie zawierał cześć syntetyczną i część krystalograficzną. Na razie jednak nadrabiam teorię i praktykę, bo obsługi dyfraktometru rentgenowskiego na studiach nie miałem, przez pierwszy semestr chodzę też na zajęcia specjalizacyjne ze studentami.
Przygotowuję powoli wpis na temat tego krystalografii i tego jak można kryształom zrobić prześwietlenie.
Mając chwilkę czasu w laboratorium, zabawiłem się w rozdzielanie na składniki czarnych markerów, jakie były na stanie pracowni do szkła i plastiku:
W jaki sposób? Techniką jaka posłużyła mi do tego zadania, była chromatografia cienkowarstwowa.
O chromatografii kiedyś już pisałem (artykuł). Jest to technika rozdzielająca mieszaniny na poszczególne składniki, pozwalająca dzięki porównaniu ze wzorcami też na ich oznaczenie. Odkryta na początku XX wieku przez rosyjskiego botanika Cwieta stała się dziś jedną z podstawowych technik analitycznych.
Cały proces opiera się o zachodzenie dwóch przeciwstawnych zjawisk - adsorpcji substancji na powierzchni chłonnego materiału i jej wypieraniu przez cząsteczki rozpuszczalnika. To na ile mocno substancja zwiąże się z podłożem zależy w dużej mierze od tego co to jest za substancja i jakie jest to podłoże.
Na adsorbencie będącym materiałem polarnym, wchłaniającym wodę, łatwiej będą się osadzać substancje polarne, hydrofilowe, zaś aby je dobrze wymyć trzeba użyć także odpowiednio silnego, polarnego rozpuszczalnika. Podobne do podobnego. Siła oddziaływania substancji z podłożem zależy od budowy i wielkości cząsteczki - obecność atomów niemetali z wolnymi parami elektronowymi (tlen, siarka, azot) sprzyja tworzeniu wiązań wodorowych, które mocniej wiążą cząsteczkę. Dla układów gdy podłoże jest niepolarne, tłuste, siłę wiązania zwiększają grupy węglowodorowe. Duża cząsteczka niepolarna może się lepiej wiązać z niepolarnym podłożem niż mała.
Natomiast siła z jaką rozpuszczalnik wymywa substancję zależy od tego jak silnie z nią oddziałuje i od tego na ile silnie wiąże się z podłożem.
Wszystkie te efekty powodują, że różne substancje mają różną siłę osadzania się na materiale chłonnym, czyli różne powinowactwo. Jeśli umieścimy mieszaninę na początku masy adsorbenta i będziemy przepuszczać przez niego rozpuszczalnik, składniki najsłabiej oddziałujące z podłożem popłyną najszybciej, a te najmocniej popłyną najwolniej. Przypomina to sytuację gdy na stadionie sportowym do biegu na kilometr zgłosi się mieszanka młodzików i dobrze wytrenowanych sportowców - ci lepsi szybko oddzielą się od słabszych, tworząc osobną grupkę.
Spróbujmy zrozumieć na czym polega to zróżnicowanie prędkości. Powierzchnia ziaren podłoża jest na tyle duża, że rozpuszczona porcja substancji nie przepływa po prostu kanalikami, tylko zostaje cała skutecznie wyłapana i osadzona. Ale zarazem z tyłu czysty rozpuszczalnik wymywa substancję i przeprowadza przez nasycone ziarna do przodu, gdzie osadza się na jeszcze nie pokrytym podłożu. Bardziej więc przypomina to ruch wydmy gnanej wiatrem niż prosty przepływ. Jeśli substancja lepiej oddziałuje z podłożem, jest słabiej wymywana przez rozpuszczalnik. W efekcie więcej czasu pozostaje związana i zostaje w tyle za lepiej wymywanymi.
W ten sposób skomplikowane mieszaniny kilkunastu czy kilkudziesięciu składników mogą zostać rozdzielone.
Barwne składniki wyciągu z zielonych liści
W moim przypadku podłożem, adsorbentem, była cienka warstwa masy krzemionkowej osadzona na aluminiowej folii. Miałem do użytku na pracowni cały arkusz, który zużywałem przy kolejnych syntezach podczas sprawdzania, czy reakcja zaszła, a gdy został mi na koniec taki nierówno wycięty kawałek, postanowiłem użyć go do opisanego tu doświadczonka.
Na starcie, nad brzegiem płytki, naniosłem kropki czterema czarnymi markerami, jakie akurat miałem dostępne. Jako naczynia użyłem najmniejszej zleweczki i przykrywki od naczynka pomiarowego. Nie pamiętam jaki dokładnie był skład rozpuszczalnika, ale generalnie był to chlorek metylenu z odrobiną octanu etylu, bo tego akurat używałem.
Aby proces chromatograficzny zachodził, należało wytworzyć ruch rozpuszczalnika w materiale płytki, użyłem tu znanego zjawiska podciągania kapilarnego - wlałem do zlewki taką ilość rozpuszczalnika, aby cały dolny brzeg był zanurzony, ale też aby zarazem same plamki mi się w nim nie moczyły, i przykryłem całość nakrywką, aby nie parowało. Zanim płytka nasiąknęła co górnego brzegu minęło kilka minut, toteż film nakręcony podczas procesu trochę przyspieszyłem:
Jak widzicie cztery z pozoru identycznie czarne plamki rozwinęły się w różnokolorowe pasma.
Generalnie rzecz biorąc nie ma czarnych barwników. Czerń powstaje wtedy, gdy substancja pochłania tak dużo światła, że oko nie rejestruje konkretnego koloru. Zwykle jednak po mocnym rozjaśnieniu czerń okazuje się być bardzo, bardzo ciemnym konkretnym kolorem. Mogą istnieć czarne pigmenty, to jest stałe substancje pochłaniające w dużym stopniu wszystkie kolory światła, tu najczęściej używany jest węgiel. Trudno jednak zastosować pigment w farbach wodnych i w flamastrach, w których tusz z wkładu przesiąka do końcówki przez porowaty materiał, działający raczej jak sito dla stałych cząstek.
Producenci używają więc mieszanek różnych barwników o dużej sile barwienia. Gdy na dany barwnik pada światło białe, pochłania on z zakresu pewne kolory a odbija inne. Jeśli dobierzemy barwniki tak, że każdy kolor będzie po trochę pochłaniany, mieszanka będzie wyglądała na czarną. A jak pokazało moje małe doświadczenie, różni producenci lubią też używać różnych, unikalnych mieszanek:
Jak widać markery Granit i BIC mają podobny składnik podstawowy - dość polarny, intensywnie fioletowy barwnik, zostający z tyłu. Zastanawiałem się czy nie jest to aby fiolet krystaliczny, ale nie miałem gencjany do porównania. Jednak dalsze składniki różnią się wyraźnie - w jednym jest to łatwo rozpuszczalny brunatny składnik, w drugim dwa składniki, jeden żółtobrązowy drugi natomiast nieco różowawy. Może być on identyczny ze składnikiem markera trzeciego "Pilot", leżącym na tej samej wysokości. Tam podstawowym barwnikiem jest leżący niżej składnik granatowy.
W przypadku czwartego markera, Pentel Pen, składniki okazały się w układzie na tyle dobrze rozpuszczalne, że bez wyraźnego oddzielenia popłynęły na sam koniec, tworząc czarną plamkę.
Ten obraz poszerzyć może badanie wyglądu płytki w ultrafiolecie, ujawniające składniki nie widoczne gołym okiem. Substancje fluoryzujące świecą własnym światłem:
Jak widzimy pojawia się nam kolejna różnica między dwoma pierwszymi markerami - BIC zawiera dodatkowy składnik świecący w ultrafiolecie na jasno niebiesko. Możliwe, że w mniejszej ilości zawierają go też dwa po bokach, słabo świecące na tej samej wysokości. Takie świecenie na brzegu kolorowej plamy oznacza, że w zastosowanym układzie rozpuszczalników nałożyły się nam na siebie dwie substancje, a więc nie udało się ich zupełnie rozdzielić.
Po co niewidoczny gołym okiem składnik w markerach? Ponieważ świeci w ultrafiolecie, to musi go też pochłaniać, jest to więc zapewne składnik chroniący pozostałe barwniki przed degradacją na świetle, powstrzymujący blaknięcie rysunków.
Różnice w składzie tuszu markerów, ale też tuszu długopisów czy atramentu piór wiecznych mają istotne znaczenie w kryminalistyce, aby wyryć czy badane dokumenty, na przykład testament, nie były później uzupełniane. Jeśli sprawca użył innego długopisu, różny skład potwierdzi dopiski. Oczywiście nie wkładamy w tym celu dokumentu do naczynia z rozpuszczalnikiem aby spojrzeć na powstające kolorowe plamki. Bądź pobiera się drobną próbkę z dokumentu i bada którąś do dokładnych technik chromatograficznych, jak wysokosprawna cieczowa, bądź wyznacza technikami nieinwazyjnymi, jak spektroskopia Ramana czy UV-Vis
A jak wykonać podobne doświadczenie u siebie w domu? Specjalistycznych płytek TLC nie trzeba kupować. Za cienki materiał chłonny wystarczy arkusz grubej bibuły, na przykład gęsty filtr do kawy, można też próbować ze sztywnym, kredowym papierem. Mi kiedyś udało się to z papierem do kserowania.
Wycinamy z papieru pasek o takiej szerokości aby zmieściły się nam kropki wszystkich flamastrów jakie chcemy zbadać, długi na kilka centymetrów. Znajdujemy wysokie naczynie o płaskim dnie, może to być słoik, szklanka, opakowanie po czymś, tak aby nasz pasek się w nim mieścił.
Teraz kwestia rozpuszczalnika - dość dobrymi, mocno wymywającymi, jest spirytus i zmywacz do lakieru do paznokci. Jeśli okażą się zbyt mocne i podczas próby wszystkie kolory od razu pójdą do góry, możemy spróbować domieszać jakiegoś słabszego składnika, może to być na przykład jakiś rozpuszczalnik do usuwania tłustych plam. Jeśli badamy markery nierozpuszczalne, pomocne może być dodanie odrobiny wody - wprawdzie jest bardzo polarna, ale gdy składniki barwne się w wodzie słabo rozpuszczają, woda może pogorszyć ich wymywanie z papieru i spowolnić. Tu już trzeba sobie poeksperymentować.
Przygotowaną mieszankę wlewamy na dno naszego naczynia, wkładamy pasek papieru z naniesionymi u dołu kropkami markerów tak aby opierał się o ściankę. Ponieważ nasiąkający papier traci sztywność, aby się nam nie przewrócił i nie wpadł możemy bądź zawinąć górny brzeg na brzegu naczynia, lub użyć spinacza do papieru, ewentualnie przewlec nitkę przez otwór w papierze i podwiązać. Pasek nie powinien przylegać do ścianki naczynia, rozpuszczalnik będzie wówczas podsiąkał w szczelinie między nimi i całość się rozmyje. Naczynie czymś przykrywamy aby rozpuszczalnik nie parował i czekamy aż cały pasek nasiąknie.
Wczoraj w laboratorium zająłem się destylacją tiofosgenu - strasznie śmierdzącego i trującego odczynnika.
Tiofosgen to formalnie rzecz biorąc podwójnie zchlorowana grupa tiokarbonylowa. Jego tlenowy analog fosgen był kiedyś używany jako bojowy gaz duszący, co daje już jakieś pojęcie o własnościach.
Zapach nie jest taki zły, jak oczekiwałem sądząc po obecności siarki. Jest ostry, drażniący a przede wszystkim duszący, podobny do innych prostych chlorków kwasowych.
Po co mi on? Do syntezy tiokarbonylodiimidazolu, a ten z kolei jest mi potrzebny do dalszych syntez. Po przeliczeniu wyszło mi, że taniej będzie otrzymać TCDI z tiofosgenu niż zamawiać gotowy. O ile uda się go w końcu zrobić bo już trzy razy próbowałem i powstawały różne dziwne produkty.