Pojawia się w zupkach chińskich, budyniach, sosach w proszku, jogurtach, kisielkach i wszelkich innych produktach wymagających zagęszczenia; tym jednak co budzi obawy nie jeśli oznaczenie E lecz rozwinięcie nazwy, mówiące o tym że jest to nie zwyczajna skrobia, ale
modyfikowana. A modyfikowane coś w jedzeniu, to na pewno jakiś szkodliwy gen - myśli niezorientowany konsument. I myli się całkowicie.
Temat GMO (genetycznie modyfikowane organizmy) budzi dziś dużo kontrowersji. Kwestii tego na ile roszczenia spierających się grup są zasadne, nie będę tutaj rozstrzygał,
niech to robią inni. Kontrowersje z tym związane są jednak często przenoszone na całkiem inne produkty i substancje, które jedynie kojarzą się z GMO. Dziś o modyfikacje podejrzewane jest wszystko co bierze się z soi, z lecytyną sojową włącznie. Na podobnej zasadzie dostaje się też modyfikowanej skrobi, której obecność w pożywieniu jest często powodem skarg na producentów. Przykład mamy
tutaj - jako produkty GMO wyliczono wszystkie ze skrobią modyfikowaną. Nieco inną wersję znajdziemy na
tym blogu - autorka wprawdzie sprawdziła, że chodzi tu o inną modyfikację, ale uważa termin za kamuflaż dla "skrobi modyfikowanej genetycznie" z której tylko i wyłącznie ma się produkować ten zagęstnik. W czym i kto nie ma racji objaśniam poniżej.
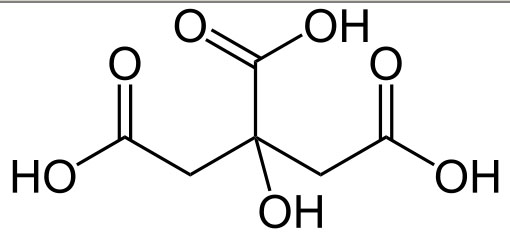
Skrobia, chemicznie rzecz ujmując, jest jednym z najpospolitszych naturalnych polimerów, stanowiąc energetyczny zapas dla roślin. Jej długie łańcuchy są zbudowane z połączonych cząsteczek glukozy:
połączonych wiązaniem α-1,4-glikozydowym. Jedna cząsteczka skrobi może się składać z od kilku setek do ponad tysiąca członów glukozowych. Zasadniczo dzieli się na dwie frakcję - amylozę składającą się wyłącznie z pojedynczych, prostych łańcuchów, i amylopektynę, której łańcuchy są w wielu miejscach rozgałęzione.
Ta różnica budowy ma istotne znaczenie dla właściwości - prosta amyloza jest nierozpuszczalna w zimnej wodzie, natomiast rozpuszcza się w gorącej. Amylopektyna częściowo rozpuszcza się w zimnej wodzie, silnie pęczniejąc i odpowiadając za kleistość mokrej skrobi.
Stanowi podstawowy składnik wielu produktów spożywczych i często jest do nich dodawana jako zagęstnik, jednak jej właściwości nie zawsze są odpowiednie. Dlatego poddaje się ją modyfikacjom, zmieniając długość i kształt cząsteczek, lub doczepiając do nich różne grupy, wpływające na zachowanie się w żywności. Jakie są to przemiany?
Najprostsza polega na częściowej hydrolizie, to jest rozdzieleniu długich łańcuchów na części, pod wpływem enzymów, podwyższonej temperatury lub kwasów. Takie kawałki nazywane
dekstrynami, mające po kilkanaście członów glukozy, są już rozpuszczalne w wodzie, tworząc lepkie roztwory i zastępując gumę arabską. Dekstryny powstają między innymi podczas wypieku chleba, odpowiadając za właściwości lekko słodkawej, chrupiącej skórki. Dekstryny są też składnikami klejów, jak choćby używany dawniej klej z prażonych kasztanów. Istnieje kilka typów dekstryn, te hydrolizowane kwasami są oznaczane jako
E 1400, hydrolizowane enzymatycznie jako
E 1405.
Inne modyfikacje polegają na potraktowaniu skrobi kwasami (
E 1401) lub zasadami (
E1402) które też rozbijają łańcuchy, ale na dłuższe cząsteczki, mające po kilkadziesiąt do stu członów. Te odmiany są już słabo rozpuszczalne w zimnej wodzie. Działając na skrobię utleniaczami, otrzymuje się s. utlenioną lub bieloną zawierającą grupy karboksylowe, zależnie od użytego utleniacza jest to
E 1403 dla wody utlenionej, i
E 1404 dla chloranu sodu. Tak zmieniona skrobia tworzy miękkie żele podobne do żelatynowych. Wszystkie te odmiany po spożyciu rozkładają się tak samo jak zwykła skrobia, tworząc glukozę.
Inne modyfikacje polegają na podstawieniu grup wodorotlenowych odpowiednimi podstawnikami. Traktując skrobię kwasem fosforowym, można uzyskać produkt częściowo podstawiony resztą fosforanową (
E 1410) lub usieciowany (
E 1412) lub usieciowany i podstawiony (
E 1413). Ta pierwsza odmiana charakteryzuje się niską skłonnością do
retrogradacji - procesu powodującego wypychanie wody z żelu skrobiowego i tworzenie zwartych agregatów o krystalicznej strukturze. Zretrogradowana skrobia jest twarda i gorzej trawiona, dla przemysłu spożywczego większe znaczenie ma jednak to, że proces retrogradacji zmienia właściwości produktu, który staje się mniej sprężysty. Odpowiada między innymi za przemiany powodujące czerstwienie pieczywa. Odmiany usieciowane tworzą żele twarde, rozpływające się w wyższych temperaturach, odporne na przemrożenie.
Jak łatwo się domyśleć, w odróżnieniu od poprzednich odmian, skrobia fosforyzowana rozkłada się z wydzieleniem reszt fosforanowych, te zaś mają już pewien wpływ na organizm. Nadmiar fosforu w takiej formie zaburza wchłanianie wapnia, zwiększając skłonność do osteoporozy. Z drugiej strony zawartość reszt fosforanowych w modyfikowanej skrobi nie może przekraczać 0,04% (ograniczenie prawne) a zazwyczaj osiąga 0,01%, trudno zatem aby doznać nadmiaru tylko tą drogą i więcej zaszkodzić może wypicie coca-coli - choć warto uwzględniać i tę ilość wobec całkowitej ilości w pozostałych produktach. Odgórnie ustalono limit zawartości w produktach dla niemowląt na 20 g/kg produktu.
Kolejne modyfikacje polegają na przyłączeniu do skrobi reszt kwasów organicznych. Jeśli potraktować ją bezwodnikiem octowym, otrzymamy skrobię acetylowaną (
E 1420) tworzącą miękkie, przezroczyste żele w niskich temperaturach. Ten podstawowy typ, ma różne odmiany, na przykład acetylowana skrobia utleniona (
E 1451), acetylowany fosforan skrobiowy (
E 1414), czy acetylowany adypinian skrobiowy (
E 1422) tworzący żele w warunkach silnie kwaśnych, podobnie do naturalnych pektyn.
Te odmiany rozkładają się z wydzieleniem kwasu octowego, który jest nieszkodliwy, jednak ze względu na możliwe działanie drażniące ustalono limit zawartości w produktach dla niemowląt na maksymalną zawartość 50g/kg produktu[
1]
Najbardziej skomplikowaną nazwę ma
E 1450 - oktenylobursztynian skrobiowy - o którym zarazem najtrudniej było mi coś znaleźć. Jest to ester w którym do pierścienia glukozy dołączono kwas zawierający dwie grupy karboksylowe i wyglądający jak pochodna kwasu bursztynowego, podstawionego oktenem - stąd zawiła nazwa (podejrzewam że równie dobrze można by go nazwać kwasem 2-acetylodek-7-enowym)
Tak duża grupa organiczna powoduje że skrobia może przylepiać się do tłuszczu na granicy faz woda-tłuszcz, stąd też używa się jej do zagęszczania majonezów, musztard i kremów. Podczas rozkładu w organizmie cała ta grupa odszczepia się i jak się wydaje, jest wydalana z moczem. Znalałem tylko jeden przegląd badań na ten temat [
2], w którym nie stwierdzono negatywnych wpływów na dwa kolejne pokolenia szczurów, których dieta zawierała nawet 30% tej modyfikacji. Jedynym negatywnym wpływem był niedobór magnezu wywołany tak ubogą dietą.
Sole glinowe tego związku są używane w pudrach do ciała i jako stabilizatory w tabletkach.
Kolejną zawiłą modyfikacją jest hydroksypropyloskrobia (
E 1440 lub
HPS) i jej kombinacje: z fosforanem (
E 1442) i z gliceryną(
E 1441). Otrzymuje się ją traktując substrat naturalny tlenkiem propylenu - epoksydem z trójkątnym mostkiem tlenowym, który łatwo się otwiera, przyłączając się do innych cząsteczek. Zależnie od stopnia podstawienia otrzymany produkt ma różne właściwości. Odmiany używane w przemyśle spożywczym zawierają do 60% podstawionych grup, tworzą żele o dobrym połysku, wytrzymałe na zamrażanie, stąd niekiedy pokrywa się nimi mrożone owoce aby spowolnić powierzchniowe utlenianie, a ponadto zagęszcza lody i ciasta mrożone. Zwykle też zastępuje się nimi mączkę chleba świętojańskiego. Stanowi też materiał rozpuszczających się w żołądku kapsułek na leki, zastępując używaną wcześniej żelatynę.
Podczas trawienia rozkłada się na glukozę i
glikol propylowy - który z kolei utlenia się do nieszkodliwego kwasu pirogronowego lub mlekowego. Mimo to wygląda na to, że może podrażniać jelita, skłaniając je do bardziej intensywnego wydalania. Krótko mówiąc może wywołać rozwolnienie. Potwierdzają to badania - u szczurów które zjadały karmę zawierającą 50% związku pojawiała się biegunka, w innym badaniu pojawiła się przy ilości 30% karmy; w badaniu na ludziach ochotnikach biegunka pojawiała się przy dawce 60 g dziennie. Zarazem nie stwierdzono negatywnych zmian w układzie krążenia, nerwowym i trawiennym u szczurów, psów i świń zjadających ten związek przez okres od kilku do kilkunastu tygodni, podobnie jak u szczurów karmionych nim przez 2 lata, a w pewnym badaniu nie wykazano negatywnych i dziedzicznych zmian u trzech kolejnych pokoleń szczurów karmionych tym związkiem.[
3] Zatem właściwie tylko biegunka przy spożyciu dużej ilości, może być uważana za jakiś wpływ zdrowotny.
Ziarna skrobi pszenicznej barwione jodem. Powiększenie ok. 500 razy
Nie takie więc te dodatki straszne, jak by to mogło wynikać z długich nazw. Dla mnie bardziej istotne jest jednak to, gdzie są one stosowane i w jakich ilościach. 30 lat temu śmietana utwardzana mąką byłaby brana za oszustwo, a jednak dziś trafiają się produkty tego typu zagęszczane modyfikowaną skrobią, i w zasadzie nikt nie widzi w tym niczego złego. Skrobia dziś zagęszcza kremy, utwardza wędliny, nabłyszcza warzywa, usztywnia mocno rozpulchnione pieczywo, a nawet zastępuje tłuszcz w produktach dietetycznych, i białko w serze do posypywania pizzy. Jest wypełniaczem w jedzeniu, a sama nie ma zbyt dużej wartości dietetycznej.
Natomiast co ze skrobią GMO? No cóż, cząsteczka skrobi nie posiada własnego DNA, zatem nie ma czegoś takiego jak "skrobia genetycznie modyfikowana" - i to skrobia i to, chemicznie cząsteczki z różnych roślin niczym się między sobą nie różnią. Nie wiem zresztą czym miałyby się różnić w przypadku pochodzenia od roślin, których modyfikacje nie dotyczyły skrobi.
Gdyby zaś materiał początkowy był zanieczyszczony na przykład białkiem roślin modyfikowanych, to wszystkie przemiany którym poddaje się skrobię, a więc traktowanie kwasami, temperaturą, bezwodnikami i innymi upochodniaczami, spowodowałyby jego rozkład, a co za tym idzie unieczynnienie. Ot i cała zagadka.
------
* Od strony technicznej modyfikowanie skrobi
wygląda tak
[1] http://www.zusatzstoffe-online.de/zusatzstoffe/313.e1420_acetylierte_st%E4rke.html
[2] http://www.inchem.org/documents/jecfa/jecmono/v17je21.htm
[3] http://www.inchem.org/documents/jecfa/jecmono/v05je70.htm