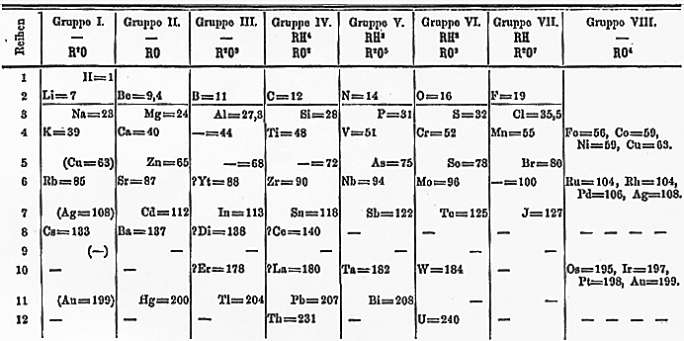
 |
| Jedna ze starych wersji tablicy Mendelejewa |
Złotym okresem odkryć pierwiastków był zdecydowanie XIX wiek, czas gdy rozwój chemii poszedł ostro do przodu, zaś kolejne pokolenia ciekawskich badaczy wsadzały do próbówek co tylko wpadło im w oko. O ile do końca XVIII wieku znano około 34 pierwiastki (chlor, odkryty w 1784 przez Scheelego początkowo uznawano za bardzo trwały tlenek) to do końca XIX wieku odkryto już 49. Natomiast lista naturalnych pierwiastków w wieku XX jest bardzo krótka.
89
Wiek otwiera pierwiastek Aktyn, którego historia była nieco skomplikowana. Nieco wcześniej, w 1899 roku chemik Andree Louis Debierne korzystając z materiałów otrzymanych z blendy smolistej, które zostały małżonkom Curie po wyizolowaniu Radu i Polonu, uzyskał z nich wysoce radioaktywną sól nieznanego pierwiastka. Ogłosił odkrycie, nadając mu nazwę aktyn, był bowiem radio-aktywny.
 |
| Świecące pod wpływem własnej promieniotwórczości sole aktynu |
Dopiero w latach 70. chemicy przyjrzeli się uważniej pierwszym doniesieniom. Aktyn Dobiernera opisany w pierwszych artykułach, miał jednak mimo wszystko inne właściwości, niż ten wyizolowany później i poddany dalszym badaniom. Po przeanalizowaniu metody otrzymania uznano, że jednak to Giesel miał rację i to on był odkrywcą pierwiastka. Nazwy jednak nie zmieniono z powrotem na Emanium, bo minęło już kilka dekad i za dużo było zmieniania.[1]
71
Lutet był jednym z ostatnich lantanowców. Jest to grupa pierwiastków, zwykle na tablicach wyrzucana pod resztę pierwiastków, ze względu na właściwości chemiczne niezwykle podobne w jej obrębie. To podobieństwo było zresztą przyczyną problemów w izolacji. Co chwila okazywało się, że zgłoszony nowy lantanowiec w rzeczywistości jest mieszanką dwóch różnych, zaś któryś z tych po wyizolowaniu i zgłoszeniu, ponownie okazywał się mieszaniną. Na końcu takiego ciągu znalazł się lutet.
Zaczęło się od badania minerałów tzw. "ziem rzadkich" nazwanych od nieczęstego występowania. Fiński mineralog John Gadolin w kopalni koło szwedzkiej wioski Ytterby odnalazł minerał, z którego wydzielił tlenek nieznanego pierwiastka. Od nazwy miejsca z którego pochodził, nazwano go Yttrium, czyli po polsku itr. Następnie po kilku dekadach jego kolega po fachu Carl Mossander odkrył, że itr zawiera domieszkę dwóch innych, podobnych pierwiastków, którym nadał niewymyślne nazwy urobione z podziału na sylaby tej pierwotnej - terb i erb. Po kolejnych dekadach Szwajcar de Marignac odkrył, że z kolei erb też nie jest czysty i wydzielił z niego kolejny pierwiastek. Będąc wierny tradycji nadał mu łudząco podobną nazwę Ytterbium, czyli itreb. Jak łatwo się domyśleć, działalność tych panów przyprawia o ból głowy studentów chemii nieorganicznej, którym wszystkie te nazwy się mylą. Natomiast mała szwedzka wioska może się pochwalić tym, że od niej nazwano aż cztery pierwiastki.
 |
| Tlenki kilku lantanowców, tzw "zemie rzadkie" |
W międzyczasie okazało się, że erb zawiera jeszcze domieszkę holmu i tulu, potem, że holm zawiera w sobie dysproz, a znany od dawna didym to mieszanka neodymu i prazeodymu. Gdy na swoje miejsce wskoczyły jeszcze gadolin, samar i europ, w tak uformowanej grupie pozostały tylko dwie irytujące dziury - pierwiastek 61 i pierwiastek 71.
Dla badaczy dość oczywistym pomysłem było szukanie wśród już znanych i wydzielonych pierwiastków. A nuż któryś okaże się mieszanką. Wreszcie w 1907 roku na trop tej samej substancji wpadli równocześnie trzej badacze - Francuz Gregore Urbain i Austriak Carl Auer von Welsbach i Amerykanin Charles James. Ten ostatni opublikował doniesienie dość późno, i nie sposób było mu stawać w szranki w boju o pierwszeństwo, natomiast pozostali panowie wszczęli kłótnię.
Urbain zaproponował dla pierwiastka nowo wydzielonego i dla oczyszczonego itrebu nazwy lutet - od zlatynizowanej nazwy regionu we Francji - i neoitreb; von Welsbach zaproponował cassiuopeium i abldebaranium, od nazw obiektów astronomicznych. Ponieważ pierwiastek nie może posiadać różnych nazw, trzeba było w końcu coś ustalić. W 1909 roku komisja zajmująca się ustalaniem dokładnej masy atomowej pierwiastków rozstrzygnęła spór, uznając że Urbain doniósł o wydzieleniu nowego pierwiastka o miesiąc wcześniej, był zatem pierwszy i może nadać pierwiastkowi nazwę.
Mimo to jeszcze do lat 50. w krajach niemieckojęzycznych używano nazwy Cassiopeium i symbolu Cp. [2]

75
Gdy losy nazwy pierwiastka 71 jeszcze się ważyły, rozpoczynało się podobne zamieszanie z pierwiastkiem 75.
Po odkryciu metody wyznaczania prawdziwej liczby atomowej i uporządkowaniu pierwiastków (zamieniono kolejnością potas i argon, które ułożone wedle masy atomowej nie bardzo pasowały do grup) okazało się, że w grupie platynowców lekkich jest jeszcze jeden pierwiastek nieodkryty. Różne grupy chemików zaczęły więc badać spektroskopowo rudy platyny.
W 1925 roku małżeństwo niemieckich chemików Ida i Otto Noddack ogłosiło wykrycie metodą spektroskopii rentgenowskiej śladów nowego pierwiastka w rudach platyny i minerale kolumbicie. Wraz z współpracownikiem Otto Bergiem wyizolowali 1 g soli tego pierwiastka, po przerobieniu 600 kg kolumbitu. Nazwali go renem, od nazwy rzeki na granicy francusko-niemieckiej.
W późniejszych badaniach ten sam zespół ogłosił wykrycie śladów pierwiastka 43, którego też brakowało w układzie okresowym, proponując dla niego nazwę Masurium, dość zresztą kontrowersyjną (pochodzi od latynizacji nazwy Mazur i w uzasadnieniu miała upamiętniać rdzennie niemiecki region). Tego wyniku nie udało się jednak powtórzyć innym badaczom.

Następnie minęło kilka dekad gdy pojawiło się doniesienie, że nawet w przypadku renu zostali uprzedzeni. W 1909 roku japoński badacz Masataka Ogawa ogłosił wykrycie pierwiastka 43 w thorianicie. Wyizolował niewielką próbkę i nazwał odkryty pierwiastek Nipponium, od jednej z nazw Japonii. Późniejsi badacze z innych krajów nie mogli powtórzyć tego odkrycia, więc nie zostało ono uznane. Dopiero w naszych czasach powrócono do oceny jego dzieł. Po przeanalizowaniu oryginalnych zdjęć z zapisem widma rentgenowskiego Nipponium stwierdzono, że wprawdzie nie zawierały pierwiastka 43, ale mogły zawierać ren, co oznaczałoby, że to Ogawa jest odkrywcą tego pierwiastka mimo błędnego przypisania. Stąd różne źródła uznają za odkrywcę renu albo Ogawę, albo Noddacków albo całą trójkę bez wdawania się w spory.[3]
72
W międzyczasie doszło natomiast do odkrycia pierwiastka 72. Pierwsze zgłoszenie w tej sprawie opublikował znany już nam Urbain przy okazji prac nad oczyszczaniem lantanowców, wydawało się bowiem, że szukany element należy do tej grupy. Ogłoszony w 1911 pierwiastek nazwał Celtium, lecz ponownie nie udało się tego odkrycia potwierdzić innymi metodami. W dużo późniejszym czasie, w związku ze sporami o pierwszeństwo, przebadano spektroskopowo próbki Urbaina, nie znajdując w nich pierwiastka 72.
 |
| Próbki metalicznego hafnu pokrytego warstewką tlenków |
W 1923 roku pracujący w Kopenhadze fizyk Niels Bohr zasugerował, że z praw okresowości można wywnioskować podobieństwo chemiczne pierwiastka 72 do znanego już cyrkonu, w związku z czym lepiej szukać w jego rudach. Sugestię podłapali chemicy Georg von Hevesy i Dirck Costler. Zbadali oni cyrkon pochodzący z norweskich rud i metodą spektroskopii rentgenowskiej wykazali ślady szukanego pierwiastka. Przy pomocy mozolnej krystalizacji frakcyjnej udało się oddzielić małą próbkę soli nowego pierwiastka. Od zlatynizowanej nazwy Kopenhagi, nazwano go hafnem.
Uznając pierwszeństwo Ogawy w odkryciu renu, hafn okazuje się ostatnim trwałym pierwiastkiem wyizolowanym z próbek naturalnych. Ale nie ostatnim w tym artykule.[4]
91
Istnienie jeszcze jednego pierwiastka między thorem i uranem było przewidywane jeszcze przez Mendelejewa. Ponieważ grupa Aktynowców nie była wtedy uznawana za odrębną, bo znano z niej tylko 2 pierwiastki, brakujący pod numerem 91 był uznawany za podobny do tantalu i w jego rudach go poszukiwano. Dlatego też wszyscy przeoczyli doniesienie Williama Crookesa, który w 1900 roku opisał wyizolowanie z soli uranu substancji, nazwanej przez niego uranem X. On sam nie opisał jej jako pierwiastka, sądził, że to raczej jakaś forma uranu nieco bardziej od niego promieniotwórcza. Na podstawie późniejszych analiz uznano, że uzyskał wtedy mieszankę thoru z pierwiastkiem 91, której dalej nie rozdzielał.
W 1913 roku Kazimierz Fajans i Oswald Göhring wyizolowali szukany pierwiastek badając produkty rozkładu promieniotwórczego uranu. Nazwali go brevium, czyli "krótkotrwały" ocenili bowiem jego czas półtrwania na zaledwie 6 godzin. Następnie w 1917 roku grupa Otto Hanna i Lisie Meitner wyizolowała z rud uranu długożyjący izotop nazwany przez nich protaktynem, bowiem rozpadał się do aktynu. W podobnym czasie na ślad pierwiastka wpadł jeszcze John Cranston, ale nie mógł opublikować odkrycia, bo powołano go na wojnę.
 |
| Protaktyn jest błyszczącym, złotawym metalem |
Zawikłany węzeł odkryć przecięła dopiero po drugiej wojnie światowej IUPAC, uznając prawo do nazwania pierwiastka dla Hanna i Meitner, ponieważ wyizolowali oni izotop o najdłuższym okresie półtrwania. Historycy nauki w związku z tym jako odkrywców uznają albo Hanna i Meitner, albo całą czwórkę z Fajansem i Goeringiem.[5]
87
Dziura w układzie okresowym na miejscu 43 została załatana sztucznie, gdy w 1934 roku bombardując neutronami molibden otrzymano technet. Dziura w miejscu 61 została załatana sztucznie wraz z otrzymaniem prometu, tymczasem tuż przed wybuchem II wojny światowej francuskiej chemiczce udało się wyizolować ostatni wyodrębniony ze źródeł naturalnych pierwiastek.
Uczennica Marii Curie Skłodowskiej, chemiczka Marguerite Perey, zajmowała się głównie pracą nad izolowaniem i oczyszczaniem aktynu z próbek lantanowców.
Pod koniec lat 30, przy pomocy precyzyjnych badań aktywności wykazała, że część próbek aktynu jest silniej promieniotwórcza od innych a zakres energii emitowanych cząstek nie pasował do izotopów aktynu. Wniosek, że zawiera dodatek czegoś silniej radioaktywnego nasuwał się sam, zwłaszcza w takiej pracowni. Wprawdzie badana substancja rozpadała się bardzo szybko, ale badając aktywność różnych roztworów, którymi przemywano próbki aktynu stwierdziła, że jest to pierwiastek o właściwościach litowców, czyli poszukiwany od dawna eka-cez.
Perey ogłosiła wyniki w 1939 roku, proponując nazwę catium i symbol Cm, w nawiązaniu do przewidywanej własności najwyższej elektrododatności. Kilka lat później jej przełożona Irena Juliot-Curie zgłosiła zastrzeżenia do takiej nazwy. Zaproponowany skrót zbiegł się ze skrótem proponowanym dla sztucznego pierwiastka kiuru, nazwanego zresztą na cześć jej matki. Ponadto anglojęzycznym chemikom catium kojarzyło się z kotami. Dość, że Perrey zaproponowała ostatecznie zmianę nazwy na francium, czyli frans, od nazwy swojego kraju i ta propozycja została w końcu zaakceptowana.[6]
 |
| 300 tysięcy atomów fransu w pułapce magnetycznej |
Był to ostatni pierwiastek, którego odkrywcy izolowali ze źródeł naturalnych. Było co prawda kilka, które otrzymano sztucznie a potem odkryto w śladowych ilościach w naturze, ale to już nie to samo.
---------
* C Fry, M Thoennessen, Discovery of the Actimium, Thoriom, Protactinium and Uranium Izotopes
[1] https://en.wikipedia.org/wiki/Actinium
[2] https://en.wikipedia.org/wiki/Lutetium
[3] https://en.wikipedia.org/wiki/Rhenium
[4] https://en.wikipedia.org/wiki/Hafnium
[5] https://en.wikipedia.org/wiki/Protactinium
[6] https://en.wikipedia.org/wiki/Francium


























-side-3D-balls.png)





