Nie wiedzieć czemu w dydaktyce szkolnej temat buforów jakoś nie jest wiązany z regułą przekory, choć przecież ich działanie jest tej zasady najlepszym przykładem. Wydaje się, że ponieważ zwykle omawia się ją przed omówieniem praw gazowych i traktuje się ją wraz z prawem działania mas jako wstęp do tego działu, stąd działanie reguły uczniowie poznają na przykładzie przemian tlenku azotu II z formy jednocząsteczkowej w dimeryczną, lub syntezy amoniaku z pierwiastków. Potem jednak nie powraca się do niej w działach dotyczących innych typów i środowisk reakcji, w efekcie wiedza zamiast się kumulować i syntetyzować, ulega podziałowi na odosobnione bloki.
Tak zwana
Reguła Przekory (ściślej zaś reguła le Chateliera-Browna), to dość arbitralne stwierdzenie, że pewne układy znajdujące się w stanie równowagi, reagują na bodźce zaburzające tą równowagę tak, jakby opierały się zmianie, dążąc do zmniejszenia skutków zmian. W tym sensie przekornie na dodatek wody reaguje układ soli na dnie naczynia z nasyconym jej roztworem - część soli się rozpuszcza i stężenie roztworu nie ulega zmianie. Zasada daje się zresztą uogólnić i na inne dziedziny, jako dotyczące również układów nie chemicznych, na przykład do przemian społecznych czy równowag ekonomicznych.
Równowaga w jakiej znajdują się układy podlegające tej regule, jest równowagą dynamiczną, co nie polega na tym, że nie zachodzą w nich żadne reakcje, lecz że reakcje przeciwstawne zachodzą z taką samą prędkością. Najlepszą ilustracją będzie tu układ naczyń połączonych, w których w prawdzie poziom wody pozostaje taki sam, lecz wskutek chaotycznych ruchów cząstek, co chwila małe porcje przedostają się z jednego naczynia do drugiego.
Jeśli podniesiemy jedno naczynie, woda będzie tak długo przepływała z niego do niższego, aż zrównoważy ją woda wpychana z powrotem. To chyba zrozumiałe. Chemicznym tego odpowiednikiem będzie na przykład równowaga między na przykład wodą, dwutlenkiem węgla i kwasem węglowym:
H2O + CO2 ⇌ H2CO3
dwutlenek węgla reaguje z wodą tworząc kwas węglowy, jednak równocześnie kwas węglowy rozkłada się na wodę i dwutlenek węgla. W normalnych warunkach równowaga jest silnie przesunięta w lewo, co oznacza że samego kwasu jest niewiele, a zresztą de facto występuje w postaci jonów jako że jego cząsteczka jest bardzo nietrwała. Jeśli zwiększymy ciśnienie gazu, tym samym zwiększając odczuwane przez roztwór stężenie (a więc ilość cząsteczek skorych do reakcji w jednostce czasu), równowaga przesunie się w prawo - część gazu się rozpuści a nadane ciśnienie opadnie. Jeśli zmniejszymy ciśnienie, bądź rozcieńczymy gaz nad roztworem na przykład azotem, część kwasu węglowego się rozpadnie i troszeczkę skompensuje spadek stężenia. Oto i przekorność.

Roztwór buforowy natomiast opiera się zmianom odczynu. Dodając kwas nie zakwasimy go do pewnych granic, dodając zasady również do pewnego stopnia go nie zalkalizujemy. Czysta przekora! Natomiast przyczyna takiego się ich zachowania jest paradoksalna - roztwory buforowe są mieszaninami kwasów z zasadami.
Żeby rzecz wyjaśnić, trzeba cofnąć się do tak podstawowej sprawy, jak definicja "kwasu" i "zasady" - a nie jest to rzecz oczywista. Pierwotnie kwasami nazywano substancje o kwaśnym smaku, zaś alkaliami nazywano produkty ługowania wodą popiołów i spalonych metali . Nieco później próbowano rzecz uściślić, przez stwierdzenie, że zasadami są substancje, które zobojętniają kwasy co jednak prowadziło do tautologii, bo dla odmiany "kwasy" definiowano jako substancje zobojętniające zasady.
Zasady, czy raczej alkalia otrzymywano z popiołów przez ługowanie wodą - skąd nazwa od arabskiego "al kali" - "popiół". Taki przesącz, nazywany potażem składał się głównie z węglanów potasu i sody z domieszką wodorotlenków. Dopiero po przereagowaniu z wapnem gaszonym (kaustyfikacji) otrzymywało się wodorotlenki, które po rozpuszczeniu w wodzie dawały "potaż żrący".
Aby nie wpaść w błędne koło w XVIII wieku ustalono ogólną definicję kwasów jako lotnych substancji zawierających niemetale, których roztwory w dużym stężeniu mają właściwości żrące i roztwarzają metale, natomiast zasadami były substancje nielotne, które reagując z kwasami usuwają ich właściwości żrące. Zasady uznano zatem za substancje będące podstawami, które zatrzymywały lotne kwasy i dawały obojętne sole i inne związki - to trochę jeszcze trącące alchemią twierdzenie przyczyniło się w pewnym stopniu do obecnego nazewnictwa. W języku angielskim, niemieckim, francuskim i w wielu innych, zasada to "baza", przy czym słowo to używane jest prawie zawsze również w niechemicznym kontekście jako "podstawa" lub "zasada działania".
Antoine Lavoisier, wybitny chemik tamtych czasów, uściślił definicję, dodając że kwasy zawierają centralny atom o wysokim stopniu utlenienia, otoczony przez atomy tlenu i powstają z tlenków niemetali, zaś z tlenków metali powstają zasady. Dla kwasów siarkowego czy fosforowego reguła się sprawdza bardzo ładnie, niestety wynika z niej, że wszystkie kwasu muszą być tlenowe. Autorytet francuskiego chemika był tak duży, że gdy
Sheele odkrył chlor, dający z czystym wodorem substancję silnie kwasową, uznano że jest to tlenek nieznanego pierwiastka, nazwanego Murium. Skoro tlen miał być pierwiastkiem charakterystycznym dla kwasów, toteż gdy przyszło nadać mu nazwę naukową podkreślono ten fakt, i z połączenia łacińskich
oxis - kwas - i
gennao - tworzyć, utworzono nazwę
oxygenium, co nasz Śniadecki, tworząc polskie nazewnictwo, próbował tłumaczyć jako Kwasoród. Na szczęście nazwa się nie przyjęła, bo już w
1810 roku
Humphry Davy udowodnił , że kwas solny, a także siarkowodorowy i selenowodorowy, nie zawierają tlenu.
Dopiero w
1838 roku
Justus von Liebig badając kwasy organiczne stwierdził, że dla kwasów charakterystyczna jest obecność wodoru, który łatwo ulega odszczepieniu i może być zastąpiony przez zawarty w zasadach atom metalu. Również w reakcji z aktywnymi metalami, jak żelazo czy cynk, wodór jest wypierany z kwasów. Ponieważ w podobny sposób w podwyższonej temperaturze aktywne metale reagują z wodą, o której było wiadome, że składa się z dwóch atomów wodoru i jednego atomu tlenu, po sprawdzeniu mas można było dojść do wniosku, o istnieniu "powodorowej pozostałości" - jonów OH. Z tą wiedzą w 1884 roku
Svante Arrhenius sformułował teorię, że kwasami są substancje, które rozpuszczone w wodzie uwalniają jony wodorowe, zaś zasadami te, które uwalniają jony hydroksylowe. Zatem do zasad i kwasów zaliczały się zarówno kwasy i wodorotlenki, jak i tlenki metali i niemetali.
Kwasy:
HCl = H + + Cl -
SO2 + H2O = 2H + + SO3-
Zasady:
NaOH = Na+ + OH-
MgO + H2O = Mg(OH)2
Niestety nie dawało się w ten sposób wyjaśnić właściwości niektórych substancji. Głównie amoniaku, który po rozpuszczeniu nie oddawał lecz odbierał wodzie wodór, uwalniając jony hydroksylowe i wywołując odczyn alkaliczny. Można tu podać jeszcze przykład kwasu borowego, który rozpuszczając się w wodzie, odbiera od niej grupę OH, pozostawiając jony wodorowe i nadając roztworowi odczyn kwaśny. Jako uzupełnienie należy dodać, że de facto wolnych jonów wodorowych się nie obserwuje - byłyby swobodnymi protonami o dosyć krótkim czasie życia - łączą się jednak z następną cząsteczką wody w jony H3O+ .
Aby trochę uzupełnić te braki, w 1923 roku dwaj badacze
Johannes Nicolaus Brønsted i
Martin Lowry, całkiem zresztą niezależnie, sformułowali inną teorię. Kwasami miały być te substancje, które oddają protony, zaś zasadami te, które je przyjmują. No tak, amoniak przyjmuje wodór, i jego roztwór jest zasadowy - jest zasadą. Wodorotlenki wiążą protony kwasów. Kwasy oddają protony. Wszystko się w sumie zgadza. Ponieważ jednak nie wszystkie rzeczy pasowały, stworzono inne teorie, aby objaśnić zachowanie się substancji w rozpuszczalnikach innych niż woda, i w stopionych solach, ale nimi nie będę się zajmował.
I cóż z tego wszystkiego wynika? Jak powiedziałem, bufory są mieszaninami kwasów i zasad. Ale Brønsteda. W roztworze są zarówno substancje oddające jak i przyjmujące protony, a więc zarówno takie, które zobojętniają kwasy, jak i te, które zobojętniają zasady. Wyobraźmy sobie, że w zlewce mamy wstawione dwie mniejsze zleweczki, w jednej kwas, na przykład siarkowy, a w drugiej zasadę, na przykład wodorotlenek sodu, i powiedzmy że będziemy przyjmować sumaryczny odczyn zlewki jako różnicę odczynów w zleweczkach. Teraz do tej z zasadą dodajmy trochę kwasu - porcja zostanie zobojętniona, zaś nadmiar zasady sprawi ze pH roztworu nie zmieni się znacząco. Albo inaczej - dodajmy trochę zasady do zleweczki z kwasem - porcja się nam zobojętni a obecny nadmiar... itp.
No ale w końcu jednak to nasze "wypadkowe pH" trochę się zmieniło, w pierwszym przypadku ubyło nam trochę kwasu a drugim zasady. Więc teraz wyobraźmy sobie, że przy pomocy magicznego zaklęcia odwracamy reakcje tak aby poziomy powróciły do wyjściowych i wszystko jest w porządku. Tym naszym magicznym zaklęciem może być tu reguła przekory, zachodzi to jednak w tych specyficznych warunkach, gdy zasada zamienić się może w kwas, a kwas w zasadę.
\>

Jak już pisałem, wedle Broensteda kwas to substancja oddająca proton. Gdy proton zostaje oddany pozostaje nam reszta kwasowa, ta jednak może równie dobrze przyjąć proton i stać się znów wyjściową cząsteczką. Skoro może przyjąć proton to jest zasadą. To nawet całkiem logiczne - gdy zobojętnimy wodorotlenek sodu kwasem solnym, powstanie nam sól kuchenna czyli chlorek sodu, gdy do jej roztworu dodamy mocnego kwasu, na przykład siarkowego, równowaga reakcji zobojętnienia - dotychczas całkowicie przesunięta w prawo - ulegnie częściowemu odwróceniu i zacznie się nam wydzielać gazowy chlorowodór. Dawniej właśnie tak produkowano kwas solny, skąd też oczywiście bierze się nazwa.
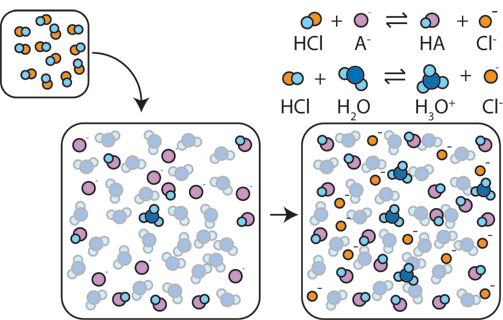
Zatem kwas solny jest kwasem, a powstający zeń anion chlorkowy zasadą. Jednak roztwór kwasu solnego i chlorku sodu nie stanowi buforu - dodanie do tej mieszanki zasady zobojętni część kwasu, sól pozostanie natomiast niezmieniona. Dodawanie kwasu też nie wywoła reakcji, przynajmniej do momentu gdy silnie kwaśne warunki nie zaczną wypierać chlorowodoru. Inaczej jest w sytuacji gdy mamy do czynienia z kwasem słabo dysocjującym.
W roztworze kwasu octowego część cząsteczek przybiera formę obojętną, CH3COOH . Cząsteczka taka może oddać proton, jest więc kwasem. Część kwasu octowego w roztworze uległa dysocjacji, a więc uwolnieniu protonu. Powstająca reszta kwasowa, anion CH3COO− może przyjąć proton, zatem jest zasadą. Oczywiście w tych warunkach zachodzi między nimi równowaga, a proton co raz jest oddawany i przyjmowany. Co jednak zwraca uwagę - przyjęcie protonu przez jon octanowy usuwa go z roztworu. W przypadku kwasu solnego jony wodorowe pochodzące od kwasu były obecne w roztworze cały czas ze względu na jego dobrą dysocjację, tutaj łączą się w obojętną cząsteczkę kwasu i nie mają wpływu na pH roztworu
Dodatek niewielkiej ilości kwasu przesuwa równowagę - część anionów octanowych przyjmie nadmiarową ilość protonów. Znikną one więc z roztworu a odczyn obniży się mniej niżby to wynikało z dodanej ilości. Dodatek niewielkiej ilości zasady zobojętni małą ilość jonów hydroniowych, mniej ich zatem będzie się łączyło z jonami octanowymi i w wyniku niezrównoważenia nieco większa ilość kwasu zdysocjuje zakwaszając roztwór, przez co podwyższenie pH będzie mniejsze niżby to wynikała z dodanej ilości zasady.
To jak duże ilości kwasów lub zasad można dodać zależy od ilości obu form w roztworze. Dla samego kwasu octowego, anionów octanowych jest mało, łatwo więc przełamać taką przekorę. Aby mechanizm mógł więc pokazać się w pełnym świetle i przeciwstawiać się większym ilościom odczynników, tworzy się mieszaniny słabych elektrolitów i ich rozpuszczalnych soli, najlepiej w jednakowej ilości.
Kwas octowy zmieszany z odpowiednią ilością octanu sodu da zatem bufor octanowy, utrzymujący odczyn w zakresie pH 3-6 zależnie od stosunku kwasu do soli. Roztwór amoniaku z dodatkiem chlorku amonu to bufor amonowy z pH 8-11. Kwas borny rozpuszczony wraz z boranem sodu to bufor boranowy, o pH 7-9.
Każdy taki roztwór zawiera stosunkowo dużo form zdysocjowanych i niezdysocjowanych, dzięki czemu może buforować odczyn zaburzany dodatkami elektrolitów w ilościach podobnego rzędu. Bufory są używane tam, gdzie potrzebny jest stały, dobrze określony odczyn, nie zaburzany zanieczyszczeniami. Wiele pomiarów właściwości substancji wykonuje się w roztworach buforujących, które utrzymują znaną wartość pH w czasie całego pomiaru. Bufory po otrzymaniu mogą być też dłuższy czas przechowywane.

Działanie buforu można wyjaśnić w jeszcze jeden obrazowy sposób - jako naczynia połączone. Jeśli wlejemy roztwór do jednego ramiona częściowo napełnionej U-rurki, to zaburzymy równowagę. Układ zachowa się jednak przekornie - część roztworu przepłynie do drugiego ramienia do momentu odzyskania równowagi. Poziom w ramieniu do którego wlewaliśmy ciecz wzrośnie zatem o połowę mniej, niż by wynikało z dodanej ilości, bo część odpłynęła. Nie można jednak czynić tego w nieskończoność, bo w końcu w rurce miejsca zabraknie, a drugie ramię nie będzie mogło tego zrekompensować bo i jemu zabraknie wolnej objętości.
Tak też jest i z buforami - mogą powstrzymywać zmiany odczynu dla ilości kwasów lub zasad mniejszych lub porównywalnych ze stężeniem własnym. Jednak z dużymi ilościami sobie nie poradzą - po całkowitym zdysocjowaniu i zobojętnieniu słabego kwasu, bufor przestaje się opierać zasadom, po całkowitym sprotonowaniu anionu reszty kwasowej, roztwór przestaje się opierać dodatkom kwasu.
Ilość odczynnika potrzebna do przełamania buforu, określamy pojemnością buforu. Zależy ona od stopnia rozcieńczenia oraz stosunku ilości formy zdysocjowanej do niezdysocjowanej.
Ot i tyle.