Częstym zadaniem na zajęciach z chemii, jest wykonanie wykresu na
podstawie wyników doświadczenia. Pomyślałem, że można by podać tu parę
przykładów, które mogą nieco pomóc zrozumieć o co chodzi, tym niespecjalnie zaznajomionym z matematyką.
Krzywa miareczkowania
Istnieje
wiele typów miareczkowań, wszystkie jednak charakteryzują się bardzo
podobnym kształtem wykresu pX od objętości dodawanego odczynnika (owym X
może być stężenie jonów wodorowych bądź innych). Wykres zależności
logarytmu zmiennej od objętości zwykle przyjmuje postać rozciągniętej
litery S, o spłaszczonych końcach i nagłym skoku wartości. Punkt końcowy
takiego miareczkowania powinien znajdować się dokładnie w połowie skoku
krzywej. Aby ją wyznaczyć należy zastosować dość skomplikowaną
konstrukcję matematyczną, którą narysowałem na wykresie z miareczkowania
potencjometrycznego, które kiedyś wykonywałem:

Należy
wybrać z prostoliniowych odcinków wykresu dwie linie równoległe i znaleźć linię do
nich prostopadłą. Od jej środka należy wyprowadzić linię prostopadłą.
Przetnie ona skok krzywej dokładnie w połowie. Powinna przynajmniej.
Można wykonywać te operacje na wydruku, linijką i cyrklem dla lepszej
dokładności. Jak jednak zauważyłem, można uzyskać ten sam efekt w inny
sposób, mianowicie każąc Excelowi narysować linię trendu. Przetnie ona
skok krzywej w połowie, jak to widać na mojej zupełnie wymyślonej
krzywej poniżej:

Sprawdzałem,
że nawet trzykrotne przedłużenie jednego z prostoliniowych odcinków
wykresu, nie przesuwa tego punktu, może to być zatem taki nieklasyczny
sposób wyznaczenie P.K.
Inny sposób to wykreślenie wykresu pochodnej.
Pochodna,
to taka funkcja, która pokazuje jak szybko zmienia się funkcja dana,
albo ściślej, jest to funkcja różnicy Y do X. W tym przypadku może to
być delta pH do czasu. Aby zrobić to w Excelu należy wpisać w jedno z
okienek taki wzór, aby uzyskać ciąg wyników odejmowań wartości pH od tej
która ją poprzedza.
Może objaśnię to na konkretnym przykładzie. Mamy taką oto tabelę danych, na podstawie której stworzyliśmy wykres ze skokiem krzywej:
Pochodna to funkcja w której zamiast kolumny wyników mamy kolumnę z różnicą między wynikiem następnym i poprzednim, więc w tym przypadku odejmujemy pierwszą wartość pH od drugiej, i wpisujemy na drugie miejsce nowej kolumny (od pierwszego wyniku nie ma od czego odjąć); na trzecim miejscu będzie wynik odejmowania drugiego wyniku od trzeciego, na czwartym trzeciego od czwartego itd. Najlepiej wpisać od razu formułę i przeciągnąć w dół:
Po czym należy stworzyć nową tabelę na podstawie otrzymanych wyników:
Jak widać dopóki odejmowane od siebie wyniki mieściły się na prostej, ich pochodna przyjmowała stałą wartość, dopiero w obrębie skoku krzywej zwiększała się. Czubek "piku" pochodnej stanowi środek skoku krzywej a więc też punkt końcowy. Dla pierwszej zmyślonej krzywej:

Jeśliby
jednak wartość skoku krzywej była mała a on sam dosyć łagodny, tak że
nasza pochodna uzyskałaby kształt mocno zaokrąglony, dla pewności można
wyprowadzić pochodną naszej pochodnej, która nam te zmiany zaostrzy.
Niestety dla niedokładnych pomiarów, w których krzywa pierwotnego
wykresu ma dosyć dużo nagłych załamań, będących punktami szybszej zmiany
wartości, wykres drugiej pochodnej może być prawie nieczytelny, jak na
tym, będącym drugą pochodną rzeczywistej krzywej z pierwszego obrazka:

Nieco inny kształt ma
krzywa dla miareczkowania konduktometrycznego. Tutaj miareczkujemy analit roztworem, zawierającym jony "niwelujące" przewodnictwo związane z jonami analitu. Na przykład miareczkowanie wodorotlenku sodu kwasem solnym.
Roztwór na początku ma dobre przewodnictwo związane z obecnością kationów sodowych ale zdecydowanie mocniej z jonami hydroksylowymi. Gdy dodamy do niego kwasu solnego jony hydroksylowe zareagują z hydroniowymi czyli zobojętnią się i powstanie słabo przewodząca woda. Wprawdzie równocześnie wprowadziliśmy aniony chlorkowe, ale ich wpływ na przewodzenie nie jest duży a całość nam się rozcieńczyła dlatego ogólne przewodnictwo spadło. W miarę dodawania kolejnych porcji kwasu przewodnictwo będzie spadało aż do punktu końcowego, czyli zobojętnienia. Po jego minięciu jony hydroniowe z kwasu będą poprawiały przewodnictwo, które będzie rosło. W dobrych warunkach otrzymamy ładny wykres w kształcie litery V jak na tym z rzeczywistego miareczkowania:
Oczywiście mówię tu o dobrych warunkach, często, zwłaszcza dla miareczkowania słabych elektrolitów, krzywa jest mniej lub bardziej wypłaszczona na dnie.
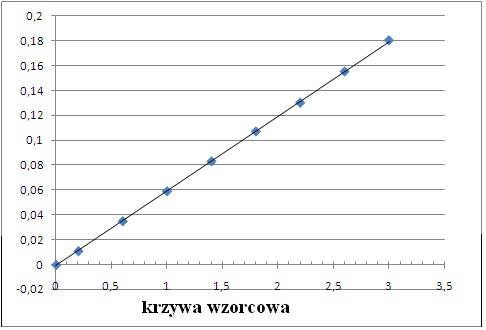
Krzywa wzorcowa
Dla
wielu przypadków nie możemy otrzymać wyniku pomiaru wprost, lecz musimy
odnosić go do pewnej skali, wykonanej w tych samych warunkach, na
przykład przez pomiar wykonany na roztworach wzorcowych o znanym
stężeniu. Z każdego pomiaru otrzymujemy jeden wynik, jeden punkt do
umieszczenia na wykresie. Teraz więc powinniśmy wyznaczyć z tego krzywą -
albo raczej prostą. Rzecz bowiem w tym że najlepiej gdy nasz wynik
mieści się w tym zakresie warunków, w którym zależność stężenie/wynik
jest liniowa, wówczas możemy bez problemu przyjąć że nawet jeśli nasz
wynik odpowiada położeniu między punktami roztworów wzorcowych, to
odpowiada stężeniu leżącemu na wyznaczonej przez nie prostej.
Jeśli wszystko przygotujemy jak trzeba a sprzęt będzie działał bez zarzutu, otrzymamy ładniutką linię jak tu:
Niestety
świat nie jest idealny a nasza dokładność ograniczona, toteż zwykle
otrzymujemy pewien zbiór punktów które nie bardzo da się na siebie
nałożyć. W dodatku wcale nie jest powiedziane, że zależność
pomiar/stężenie musi być liniowa dla wszystkich warunków. Weźmy na
przykład taki wykres zależności absorbancja/stężenie dla pewnego
kompleksu:
Dla
niskich stężeń kompleks jest mało trwały i zabarwienie jest słabsze niż
by to wynikało z rozcieńczenia; po przekroczeniu pewnej granicy
zależność jest liniowa i daje się opisać pewnym wzorem matematycznym,
natomiast powyżej pewnego stężenia jony kompleksu tworzą konglomeraty
zabarwione silniej. W dodatku w roztworze nie zawierającym kompleksu
przyrząd zmierzył jakaś absorbancję i wykres nie zaczyna się w punkcie
0/0. Tak się akurat może zdarzyć i zdarzało się, chociaż powyższy wykres
został wymyślony.
Teraz próbujemy za pomocą tego wykresu wyznaczyć z absorbancji trzech próbek ich prawdziwe stężenia - dla punktu
1 jest to łatwe, bo nałożył się na jeden z punktów pomiarowych, w tym przypadku odpowiadając stężeniu 0,09. Dla punktu
2 też nie jest to trudne - zmieścił się w zakresie zaznaczonego na czerwono odcinka liniowego, możemy więc wyznaczyć właściwe stężenie metodą graficzną, zaznaczając punkt na wykresie i sprawdzając jakiemu stężeniu odpowiada (powiedzmy że 0,055), możemy też jednak zrobić rzecz dokładniej.
Możemy wyciąć z wpisanej w Excela tabeli kawałek, odpowiadający liniowej zależności i po stworzeniu na jego podstawie wykresu kazać programowi aby wyrysował linię trendu z równaniem określającym zależność X od Y - w tym przypadku równanie miało postać
Y = 8,109x + 0,928. Wiedząc jakie jest Y (absorbancja) możemy wyliczyć X czyli stężenie i równanie będzie pasowało dla wszystkich pomiarów mieszczących się w tym zakresie.
Pozostał nam jednak jeszcze jeden pomiar, który wypadł poza odcinkiem liniowym, w dodatku między punktami pomiarowymi. Możemy wykreślić między nimi odcinek i założyć, że gdy postawimy nasz punkt na tej linii to będziemy mogli odczytać prawidłową wartość, ale któż mógłby nam zaręczyć, że akurat na tym odcinku prawdziwa zależność nie wije się wedle pewnej nieprostej linii, nie poddającej się opisowi przez równania? Na przykład takiej oznaczonej zieloną krzywą. Nikt nie może czegoś takiego przewidzieć, a jeśli nie wiemy dokładnie jaka jest tutaj zależność i jakiemu Y odpowiada jaki X to nie wyznaczymy zależności ani graficznie ani matematycznie zaś wynik który możemy otrzymać będzie obarczony błędem, który może być największym błędem w całej analizie. Dlatego właśnie najlepiej wyznaczyć krzywą przed właściwym badaniem i starać się aby stężenia analizowanych próbek w miarę możliwości mieściły się w odpowiednim zakresie.
Wprowadźmy jednak nieco więcej chaosu. Może się zdarzyć że nasz sprzęt jest stary i zdarzają mu się duże szumy, zafałszowujące wyniki w obie strony i gdy naniesiemy punkty na wykres otrzymamy taką rozsypkę:
No cóż, bywa i tak. Jeśli jednak faktycznie są to przypadkowe szumy, to pamiętajmy że przypadek szumi średnio statystycznie po równo w obie strony - zatem w tym obłoczku jest prawdziwa linia trendu, a tylko złośliwie ktoś po przesuwał część punktów nad i pod nią. W takiej sytuacji musimy albo zdać się na opcję "dodaj linię trendu" albo samemu wyznaczyć jakąś linię, która znajdzie się najbardziej pomiędzy punktami. Dobra linia powinna przechodzić przez możliwie najwięcej punktów oraz umiejscawiać się pośrodku między skrajami grupy, a także mieć w miarę możliwości tyle samo punktów nad co pod nią, jak w tym przypadku:
gdzie linia przechodzi wprawdzie tylko przez trzy punkty, ale ma równo po cztery nad sobą i pod.
Ale oczywiście - powiecie zaraz, chyba łatwiej jest zlecić to Excelowi? Jak najbardziej, tylko że czasem rzucając mu surowe dane otrzymamy linię trendu zupełnie fantazyjną, jak na poniższym przykładzie:
Zdawałoby się że wyszłaby nam ładna linijka z małymi odchyleniami, ale jeden punkt ma wartość prawie dwa razy za dużą, dlaczego? A bo na przykład obluzował się kabelek przy elektrodzie, albo kuweta kolorymetru nie była przetarta i na drodze wiązki znalazła się rozpraszająca kropla. Tak się może zdarzyć, jeśli zauważymy rzecz w porę, będziemy mogli powtórzyć pomiar tej próbki, jeśli tego nie zrobimy pozostaniemy z bardzo dziwnym wynikiem.
Jedną z rzeczy które prowadzący bardzo tępili było takie właśnie bezrefleksyjne działanie, ślepe zawierzenie procedurze, jakie obrazuje powyższy wykres. Nie trzeba wielkiego doświadczenia aby zauważyć, że z linią trendu jest coś nie tak. Wprawdzie program wyrysował ją poprawnie i zgodnie z zasadą "najbardziej wypośrodkowanej linii" ale przecież leży na niej tylko jeden punkt a zupełnie pominięto kilka leżących na wyraźnej prostej. Niestety bywało (a nawet i mi dawniej się przydarzało) że uczeń zgodnie z procedurą w ćwiczeniu tworzył wykres, kazał rysować programowi absurdalny trend, potem równanie i z równania wychodziły mu wyniki z błędem rzędu 150% i więcej. I co teraz z tym zrobić?
Do śmieci. Jeśli widzimy że wynik jest absurdalnie za duży lub za mały i występuje sam jeden dziwak wśród innych normalnych, to pomińmy go jako błąd i wyrysujmy wykres dla pozostałych danych, z czego na przykład otrzymamy coś takiego:
I od razu lepiej. Z reguły w pomiarach posiadających bardzo dużo punktów pomiarowych takie przypadkowe szumy można eliminować odpowiednimi programami, należy jedynie określić jakiś przedział, w ramach którego wyniki uznaje się za błąd, na przykład zależność powinna być liniowa i punkty odbiegające od sąsiednich - poprzedniego i następnego - o więcej niż powiedzmy 5% są eliminowane, zaś te z mniejszym odchyleniem włączane i uwzględniane. W ten sposób wykres się nam wygładza a błąd zmniejsza.
Praktyka ta ma jednak swoją złą stronę - pod pozorem eliminowania punktów błędnych nieuczciwi badacze (i studenci) mogą dopasować dane do z góry powziętej tezy. Jeśli ktoś chce wykazać silną zależność na przykład między dawką leku a spadkiem odczuwalnego stopnia bólu głowy, może wyeliminować z części wykresu dolne punkty, z drugiej górne i prowadząc trend po punktach skrajnych wykazać silne działanie preparatu. Zwykle wykrywa się takie oszustwa przez dokładną analizę - jeśli na przykład w badaniu wykazano silną korelację między podlewaniem krzewów kawowych krowim moczem a spadkiem aflatoksyn w ziarnach, lecz zarazem jako błędne wyeliminowano 40% danych, to coś tu jest nie tak.
Może coś to pomoże.
ps. ponieważ ostatnio ten post miał spore zainteresowanie, dodałem praktyczny przykład postępowania.