
W 1992 roku pewna belgijska klinika na obrzeżach Brukseli, specjalizująca się w leczeniu otyłości, postanowiła poszerzyć swoją ofertę o środki bardziej egzotyczne i w oferowanej kuracji zaczęła stosować mieszankę chińskich ziół, którym tradycja przypisywała wspomaganie odchudzania. Po kilku miesiącach u pacjentek zaczęły się jednak pojawiać niepokojące objawy niewydolności nerek. U części doszło do niebezpiecznych dla życia martwic i zwłóknienia, potrzebne były przeszczepy. Szybko powiązano choroby nerek ze stosowaniem ziół, co jednak sprawiało ten problem, że składniki sprowadzanej mieszanki, a więc liść magnolii i ziele stefanii (Stephania tetrandra) nie były toksyczne dla nerek. Zatem powodem musiał być jakiś dodatkowy czynnik chorobotwórczy.
Tymczasem liczba chorych wzrastała i były to też osoby spoza pacjentów kliniki, które używały tej samej mieszanki. Na podstawie powtarzalności objawów uknuto termin medyczny "nefropatia chińskich ziół". Dopiero wraz z rozwojem technik analitycznych udało się w 1994 roku zidentyfikować w stosowanym preparacie prawdopodobną substancję toksyczną - był to kwas arystolochowy, występujący obficie w roślinach z rodzaju Kokornak. Spośród których kilka gatunków jest używanych w tradycyjnej medycynie chińskiej...
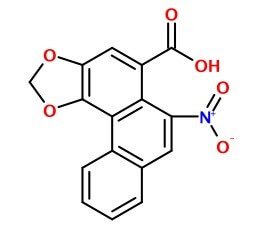
W późniejszym śledztwie wyszło na jaw, że pojawienie się go w mieszance było prawdopodobnie wynikiem pomyłki. Chińska nazwa suszu stefanii to Han Fang Ji, gatunek kokornaka Aristolochia fangchii używany w innych schorzeniach to Guang Fang Ji. Zamówienie z Belgii podawało nazwę mało precyzyjnie jako Fang Ji. W tej sytuacji nie trudno o pomyłkę.

Kokornaki to rośliny zielne, lub drewniejące, zazwyczaj pnącza lub krzewinki o sercowatych liściach i rurkowatych kwiatach, niekiedy pułapkowych. Wiele gatunków jest uprawianych jako rośliny ozdobne i okrywowe. W Europie naturalnie rośnie kilka gatunków, spośród których najpospolitszy jest rosnący też w Polsce kokornak powojnikowy (Aristolocha clematitis).
W dawnej medycynie europejskiej kokornak był stosowany jako lek na schorzenia wątroby, przy żółtaczce, jako środek poronny lub przyspieszający poród, czy wewnętrznie na rany.[2] W tradycyjnej medycynie chińskiej wykorzystywanych jest kilkadziesiąt gatunków, zwykle były używane przy zapaleniu stawów i obrzękach, niektóre gatunki jako środki przeciwpasożytnicze. Pewien gatunek jest uważany za środek do odstraszania węży, w związku ze specyficznym, nieprzyjemnym zapachem wielu kokornaków.
W związku z tym odnotowane zatrucia w Belgii, których do 1994 roku doliczono się 105, wydają się być zaskakujące - jeśli od starożytności ziół tych używano w Chinach i Europie, to czemu wcześniej nie spostrzeżono ich toksycznego działania? Przyczyną jest zapewne czas stosowania - w dawnych zastosowaniach kokornaki były używane doraźnie, przez krótki czas. W tym przypadku ziołowy suplement na odchudzanie był zażywany regularnie przez kilka miesięcy, przez co objawy pojawiły się na tyle szybko, że można było je powiązać z użyciem suplementu.
Wyglądałoby zatem na to, że spożycie sporadyczne nie powinno być niebezpieczne.
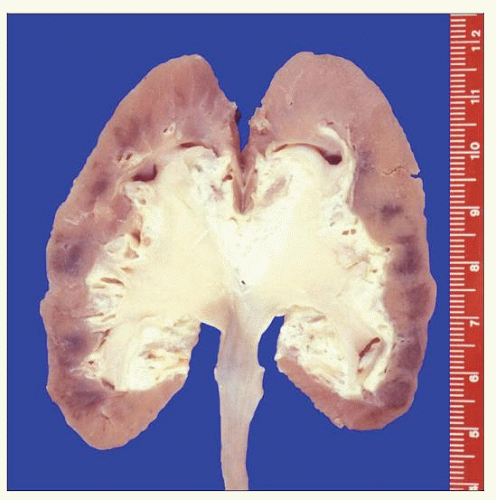
Wiele badań wskazuje też na bardzo silne działanie rakotwórcze kokornaków. W jednej z prac [3] opisano wyniki badań nerek i moczowodów usuniętych w związku z martwicą wywołaną kwasem aristolocholowym. Na 39 pacjentów u połowy w usuniętych nerkach stwierdzono ogniska nowotworów, u pozostałych pojawiały się dysplazje nabłonka będące stanem przedrakowym; żadnych zmian nie miały tylko dwie osoby.
Kwas arystolochowy ze względu na budowę, a jest płaską cząsteczką aromatyczną, ma skłonność do tworzenia interkalacji z DNA. Wpasowuje się pomiędzy płaskie cząsteczki zasad purynowych w nici kwasów nukleinowych i zaburza ekspresję genów. Podczas podziałów komórkowych generuje też punktowe mutacje związane z nieprawidłowym odczytem kodu. Wśród z genów które ze względu na położenie w chromosomach są przezeń blokowane częściej, jest też TP53, odpowiedzialny za wytwarzanie białka hamującego nowotworzenie. Mutacje w tym genie zostały uznane za najbardziej charakterystyczny skutek działania kwasu aristolocholowego. Ponieważ toksyna jest szybko wydalana przez nerki i zagęszcza się w moczu, zmiany mutagenne dotyczą najczęściej komórek nerek i nabłonka przewodów moczowych.
Połączenia DNA-AA (aristocholic acid) są bardzo trwałe, udawało się je znaleźć w komórkach nerek pacjentów z nowotworami, którzy zażywali kokornak kilkanaście lat wcześniej.
Gdy pojawiły się publikacje łączące charakterystyczne uszkodzenia nerek z narażeniem na kwas arystolochowy, zauważono podobieństwo objawów do znanej już od dawna Endemicznej nefropatii bałkańskiej. Chorobę opisano po raz pierwszy w latach 20. jako specyficzną dla pewnych społeczności w dolinie Dunaju i dopływów, na terenach obecnej Chorwacji, Bośni, Serbii, Rumunii i Bułgarii. Szczególnie dużo przypadków występowało w okolicach miasta Wraca.
Choroba rozwijała się wolno, występowała tylko u dorosłych w wielu 30-60 lat. Późniejsze badania u emigrantów którzy wyjechali z regionu endemicznego pokazały, że warunkiem zachorowania jest przybywanie w tamtej okolicy przez minimum 20 lat. Choroba przybierała postać przewlekłego, śródmiąższowego zapalenia nerek i stopniowo doprowadzała do zwłóknienia i martwicy, wymagających usunięcia narządu i dializowania lub przeszczepów. W sytuacji raczej kiepskiej opieki medycznej na tamtych terenach często nefropatia doprowadzała do przedwczesnych zgonów.
W średnio 50% przypadków nefropatii towarzyszyły nowotwory, głównie rak nabłonkowy nerek i przewodu moczowego. Szacuje się, że nawet w naszych latach symptomy o różnym nasileniu posiada co najmniej 25 tysięcy osób.

Przez długi czas podawano różne możliwe przyczyny endemicznego występowania tej choroby. Występowała wyraźnie rodzinnie ale nie była wprost dziedziczna. Pojawiała się u osób z innych rejonów, które mieszkały w regionie endemicznym dostatecznie długo; pojawiała się u mieszkańców rejonu endemicznego którzy przeprowadzili się w inne miejsce. Obszar występowania bałkańskiej nefropatii od kilku dekad pozostaje taki sam - nie pojawiły się nowe ogniska, ani nie zaniknęły stare. Próbowano więc powiązać ją z czynnikami lokalnymi.
Zauważano na przykład, że podstawowym pożywieniem w tej okolicy są zboża, zaś większość chorych było rolnikami, przy czym ze względu na klimat i zwyczaje ziarno często zanieczyszczone było pleśnią, stąd też prawdopodobne wydawało się iż znaczenie ma tu jakaś toksyna. Najbardziej prawdopodobna wydawała się Ochratoksyna A, wytwarzana przez pleśnie, której obecność w paszy wywołuje w krajach północnej europy nefropatię u świń. Inna hipoteza skupiała się raczej na zbieżności obszaru zachorowań z obszarami wydobycia węgla, sugerując jakiś wpływ metali ciężkich z wód pokopalnianych czy niedoboru selenu w glebach nad złożami.
Jednak w latach 90. zauważono, że objawy nefropatii bałkańskiej i nefropatii ziół chińskich są do siebie zaskakująco podobne. Kokornak jest na tamtym obszarze bardzo pospolity, stanowi częsty chwast polny i części rośliny lub nasiona mogą zanieczyszczać zboża.
Hipoteza ta nie do końca tłumaczy wszystkie własności choroby, zwłaszcza silny endemizm nieraz ograniczający się do pojedynczych gospodarstw we wioskach, wydaje się więc, że nakłada się tutaj wiele przyczyn - sporadyczna ekspozycja na kokornak, niedobory w diecie, tryb życia i czynniki genetyczne. Ostatnio opublikowana praca na ten temat wskazuje na ten ostatni czynnik - alterację genów na chromosomie 3 w miejscu 3q25-26. Posiadacze tej mutacji są wyjątkowo wrażliwi na działanie toksyny kokornaku i to u nich rozwija się choroba. Tłumaczy to dlaczego spośród osób z rejonu endemicznego choruje tylko około 8% mieszkańców. Widocznie wrażliwość ta ma też różne natężenie, u nieszczęsnych mieszkańców południowej Europy rzadka ale powtarzalna ekspozycja na kokornak wywoływała objawy u kilku procent; wśród pacjentek belgijskiej kliniki, które regularnie łykały zioło przez kilka miesięcy, objawy rozwijały się u nawet 20%.[4]
Addukty DNA-AA zostały też wykryte w usuniętych organach, potwierdzając, że chorzy musieli być narażeni na ten związek.
Tymczasem pojawiają się kolejne doniesienia. Artykuł z Tajwanu przekazuje wyniki badań populacyjnych w których badano jaka jest częstość narażenia chorych na nowotwory na różne czynniki toksyczne, w porównaniu z resztą populacji. Okazało się, że narażenie na medykamenty zawierające azjatyckie gatunki kokornaków zdarzało się takim pacjentom wyraźnie częściej. Związek statystyczny okazał się silniejszy niż nawet narażenie na dym papierosowy. [5]
Znalazłem także opis polskiego przypadku nefropatii powiązanego z użyciem takiego preparatu. 17-letni pacjent z wyraźną nadwagą zgłosił się w związku z bólami i zawrotami głowy, których doznał w trakcie kuracji odchudzającej, stwierdzono u niego nadciśnienie. Po upływie kilku miesięcy nadciśnienie utrzymywało się, a do objawów doszedł świąd, nudności i osłabienie. Tym razem stwierdzono u niego białkomocz. Rozpoznano u niego przewlekłą nefropatię cewkowo-śródmiąszową i wtórną kwasicę. W ciągu następnych miesięcy choroba rozwinęła się tak bardzo, że konieczny był przeszczep nerki. W międzyczasie okazało się, że chory zażywał ziołowy preparat mający zawierać w składzie tylko niegroźne rośliny, jak rozmaryn, jeżogłowkę, tymianek i żeńszeń, który jednak podczas badań laboratoryjnych okazał się zawierać też kwas arystolochowy.[6]
W 2010 roku w Wielkiej Brytanii skazano właścicielkę chińskiej zielarni, która sprzedawała preparat Xie Gan Wan zawierający kokornak. Zażywająca pigułki 58-letnia kobieta, której miały pomóc na problemy dermatologiczne, doznała uszkodzenia nerek i raka dróg moczowych.[7]
Z tych powodów zaleca się obecnie, aby preparatów ziołowych zawierających kokornaki nie stosować nawet incydentalnie, a także unikać mieszanek zawierających ziele Stefanii, ze względu na możliwą pomyłkę nazwy rośliny u chińskiego producenta. Chińskie nazwy[8] gatunków kokornaka używanych w niektórych mieszankach to:
- Guang Fang Ji (Fangchi) - Aristolochia fangchi
- Xixin - Radix et Rhizoma Asari
- Guan Mu Tong - Aristolochia manshuriensis
- Qing Mu Xiang - Aristolochia cucurbitifolia
- Ma Dou Ling - Aristolochia debilis
- Tian Xian Teng - Aristolochia contorta
--------
Źródła:
[1] https://link.springer.com/article/10.1007/BF00302751
[2] http://rozanski.li/?p=824
[3] L. Nortier et al.; Urothelial Carcinoma Associated with the Use of a Chinese Herb (Aristolochia fangchi), N Engl J Med 2000; 342:1686-1692June 8, 2000 DOI: 10.1056/NEJM200006083422301
[4] Marie Stiborová, Volker M. Arlt, and Heinz H. Schmeiser Balkan endemic nephropathy: an update on its aetiology, Arch Toxicol. 2016; 90(11): 2595–2615.
Published online 2016 Aug 19. doi: 10.1007/s00204-016-1819-3
[5] Hsiao-Yu Yang, Pau-Chung Chen, and Jung-Der Wang, Chinese Herbs Containing Aristolochic Acid Associated with Renal Failure and Urothelial Carcinoma: A Review from Epidemiologic Observations to Causal Inference, BioMed Research InternationalVolume 2014 (2014), Article ID 569325, 9 pages http://dx.doi.org/10.1155/2014/569325
[5] Hsiao-Yu Yang, Pau-Chung Chen, and Jung-Der Wang, Chinese Herbs Containing Aristolochic Acid Associated with Renal Failure and Urothelial Carcinoma: A Review from Epidemiologic Observations to Causal Inference, BioMed Research InternationalVolume 2014 (2014), Article ID 569325, 9 pages http://dx.doi.org/10.1155/2014/569325
[6] Konrad Walczak, Anna Krysicka, Dariusz Moczulski, Nefropatia ziół chińskich — opis przypadku, Forum Nefrologiczne 2010, tom 3, nr 4, 272–276 (PDF)
[7] http://wiadomosci.onet.pl/kiosk/leczenie-wysokiego-ryzyka/6tbhb
[7] https://www.hindawi.com/journals/bmri/2014/569325/tab1/
[7] http://wiadomosci.onet.pl/kiosk/leczenie-wysokiego-ryzyka/6tbhb
[7] https://www.hindawi.com/journals/bmri/2014/569325/tab1/


























.jpg)

