Miał to być kolejny szczęśliwy dzień. Lakhvinder Cheema wrócił z pracy do swojego domu w Feltham, zachodniej dzielnicy Londynu i odgrzewał potrawkę z kurczaka w sosie curry, zagadując do swojej narzeczonej Gurjeet. A mieli dużo do obgadania, zaledwie za dwa tygodnie, w Walentynki, zamierzali wziąć ślub, który przy okazji zalegalizuje pobyt narzeczonej w Anglii. Znali się dopiero od kilku miesięcy, ale byli zupełnie zdecydowani aby przeżyć ze sobą następne lata.
Potrawka podlana ostrym sosem smakowała mu tak bardzo, że zjadł podwójną porcję. Jednak gdy już kończył w znajome pieczenie w ustach wkradło się nietypowe wrażenie drętwienia i mrowienia. Zdziwiony dotknął palcem języka i ust stwierdzając, że zupełnie nic nie czuje.
- Źle się czuję - powiedział do narzeczonej. - To chyba coś w jedzeniu.
Próbował wstać, ale nogi się pod nim załamały. Gurjeet nie mogła mu pomóc, bo sama znalazła się w podobnym stanie. Poczuła zawroty głowy a światło w kuchni osłabło aż do zupełnych ciemności. Przezwyciężając osłabienie Cheema zadzwonił na pogotowie, dodając ostatkiem sił:
- Ktoś dodał truciznę do jedzenia. To moja była dziewczyna.
Zaledwie kilka minut później oboje byli już bezwładni a ich ciałami wstrząsały drgawki. Godzinę po przyjęciu do szpitala, Cheema zmarł, a Gurjeet musiała zostać podłączona do respiratora. Najważniejszym było teraz dociec jaka substancja wywołała tak gwałtowne skutki.
Jeszcze tego samego dnia policja zatrzymała Lakhvir Singh, byłą kochankę Cheema. Podczas przeszukania znaleziono przy niej torebeczkę z drobnym, roślinnym pyłem.
- To proszek na wysypkę - tłumaczyła. Początkowo jednak nie zbadano tego tropu. Objawy były tak gwałtowne, że sądzono, że to zatrucie cyjankiem potasu.
Singh poznała Lakhvindera dawno temu. Uwikłana w niezbyt szczęśliwe małżeństwo, możliwe że aranżowane, szybko zwróciła uwagę na młodego Sikha który zaczął wynajmować u nich pokój. Był świeżo po rozwodzie, samotny, nic więc dziwnego, że między nim a żoną najemcy zawiązała się bliższa zażyłość. Nawet gdy znalazł sobie inne mieszkanie i tak przychodziła do niego niemal codziennie, gotowała, sprzątała i sypiała z nim jak prawdziwa żona. Gdy mąż Singh zachorował na raka i musiał przejść długotrwałe leczenie, uznała to za dobrą okazję aby częściej bywać z kochankiem. W trakcie trwającego 16 lat romansu dwa razy zaszła w ciążę, którą na jego prośbę usunęła.
Ale w miarę upływu lat Cheema starał się od niej oddalić, będąc zmęczony jej zaborczym podejściem. W 2008 roku krewni zaczęli swatać go z o połowę młodszą Gurjeet Choongh, która niedawno przyjechała do Anglii. Od razu się sobie spodobali i szybko formalnie się zaręczyli. Odrzucona kochanka szalała ze złości, wysyłając mu nienawistne wiadomości. Mimo to jeszcze przez pewien czas przychodziła do jego domu, aż na początku grudnia Cheema powiedział dość.
 |
| Lakvinder Cheema i jego narzeczona Gurjeet Choongh |
Zbadanie rodzaju trucizny pomimo zabezpieczenia proszku z kieszeni podejrzanej, porcji nie zjedzonego sosu curry i wymiocin ofiar, okazało się jednak dużo trudniejsze niż się to początkowo wydawało. Cyjanek szybko wykluczono. Ze względu na objawy sugerujące porażenie nerwów, brano pod uwagę neurotoksyczne pestycydy, ale mogły być to też trucizny roślinne. Trudno było choćby przybliżyć rodzaj substancji, dlatego analitycy postanowili użyć metody najbardziej ogólnej, która powinna wykrywać i zidentyfikować niemal wszystko - analizy chromatograficznej sprzężonej ze spektrometrią mas.
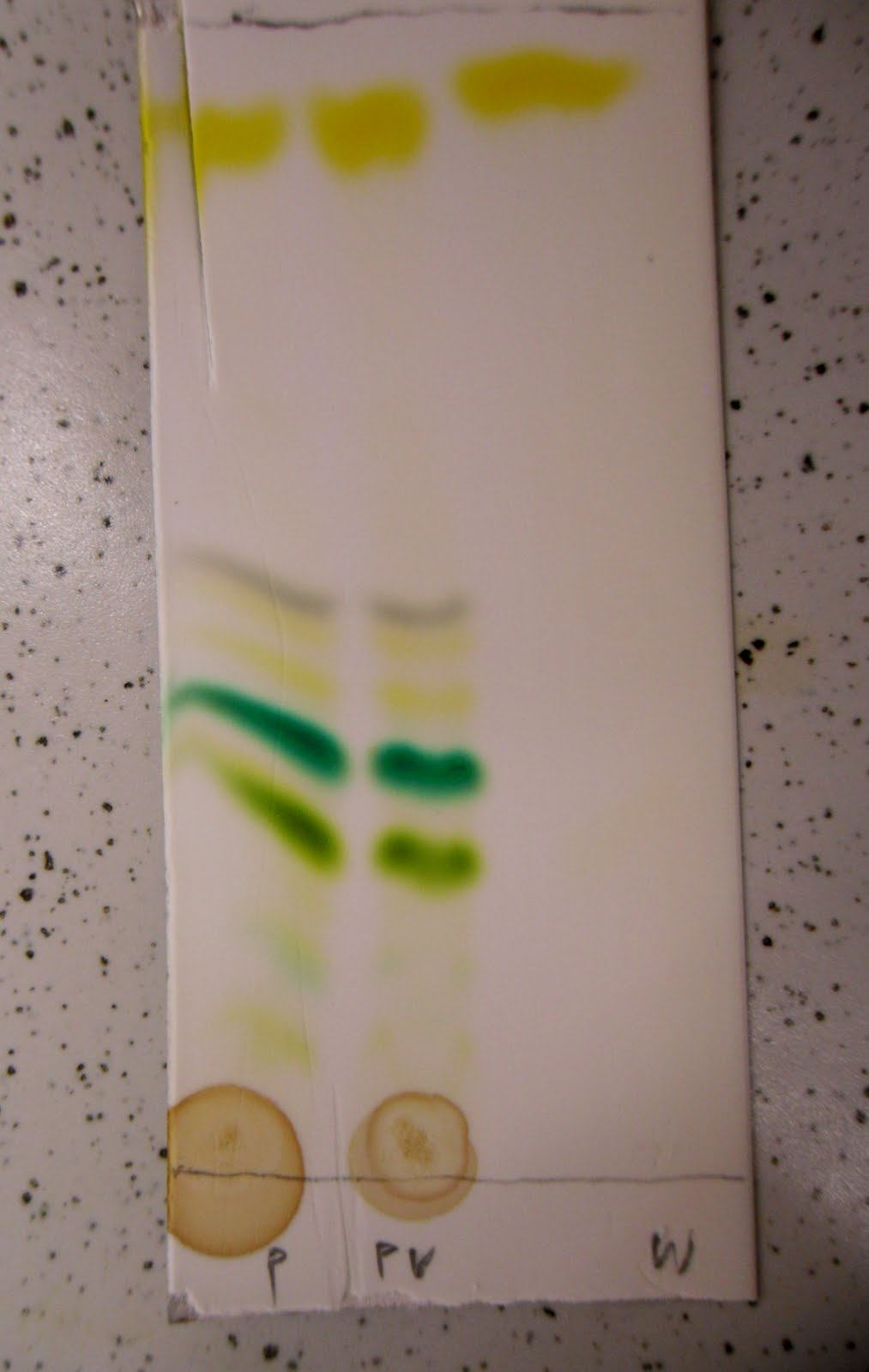
Chromatografia to metoda rozdziału mieszanin oparta o różne powinowactwa składników mieszaniny do adsorbentu wypełniającego długi przewód lub kolumnę. Każda substancja, której roztwór lub opary są przepuszczane przez dobrze adsorbujący, porowaty materiał, w jakimś stopniu oddziałuje z jego powierzchnią. Siła tego oddziaływania zależy w dużym stopniu od tego jaka to jest cząsteczka, a więc jakie ma grupy funkcyjne, jaki ma moment dipolowy, hydrofilowość, rozmiar cząsteczki itp. Jedne będą więc mocniej się wiązały z wypełnieniem a inne słabiej. Podczas przepuszczania porcji mieszaniny substancji przez kolumnę z odpowiednim wypełnieniem, te najsłabiej z nim oddziałujące substancje będą najefektywniej wymywane przez rozpuszczalnik, wypierane przez mocniej oddziałujące i w efekcie oddzielą się jako osobna porcja, podróżująca przez kolumnę szybciej, na przedzie.
 |
| Chromatografia wyciągu z zielonego liścia na płytce pokrytej warstwą adsorbentu. Od góry: karoten, feofityna, karotenoidy, chlorofil a i b |
Sumą tych wszystkich procesów powinno być zatem pooddzielanie składników mieszaniny i możliwość zbadania czystych. Jednakowoż oddzielenie i oczyszczenie składnika mieszaniny, niczego nam o nim nie mówi, dlatego dla identyfikacji wykorzystuje się inny skutek wyżej opisanych procesów - dla danej substancji w danych warunkach czas przejścia przez kolumnę z określonym rodzajem wypełnienia jest taki sam.
Jeśli więc najpierw przepuścimy przez kolumnę mieszaninę, rejestrując detektorem pojawianie się kolejnych składników i czasy ich przebiegu, a potem, nie zmieniając warunków, małe ilości wzorców substancji których obecności się w mieszaninie spodziewamy, to stwierdzenie, że pewne składniki miały identyczny czas przebycia co dany wzorzec jest równoznaczne z potwierdzeniem, że są to te same substancje.
No dobrze. A co w przypadku gdy nie wiemy nawet jakiego użyć wzorca, bo morderczyni użyła trudno powiedzieć jakiej trucizny? Tutaj z pomocą przychodzi dołączenie do zestawu chromatograficznego detektora, który prócz wykrycia, że dotarł do niego rozdzielony składnik mieszaniny, może nam coś o nim powiedzieć - analizatora przy spektrometrii mas.
Spektrometria mas to metoda bardziej bezpośredniego badania składu substancji. Zasada działania jest generalnie bardzo prosta - jeśli zjonizujemy badaną substancję i poddamy otrzymane jony działaniu pola elektrycznego, to będą się w nim poruszać nabierając pewnego pędu. Jony o tym samym ładunku ale różnej masie w tym samym polu zyskają różny pęd. Możliwe jest zbudowanie takiego układu, w którym następować będzie odsiewanie jonów o niepasującej masie. Może to być na przykład rurka skręcająca pod ostrym kątem, w której układ magnesów tworzy kanał zaginający jony. Jony lekkie będą zbyt silnie zaginane i uderzą w ściankę, te zbyt ciężkie nie będą się wyrabiały na zakręcie i też uderzą w ściankę. Zmieniając moc magnesów możemy zmieniać optymalny zakres mas w którym jony są w stanie przejść przez układ filtrujący. W efekcie z wiązki jonów najpierw zarejestrowane zostaną te o jednej masie a potem te o innej.
Wiedząc jak zmienialiśmy parametry układu filtrującego możemy zatem określić jaką dokładnie masę cząsteczkową musiały mieć jony które w danym momencie, po przejściu przez układ uderzyły w detektor i zostały wykryte. Szybkie przemiatanie ustawieniami analizatora po zjonizowaniu próbki da nam widmo masowe jonów powstałych z danej substancji, a to jest już w stanie powiedzieć nam coś o tym z jaką konkretnie substancją mamy do czynienia.
Zależnie od metody i siły jonizacji analizuje się bądź masy jonów pojedynczych cząsteczek, bądź masy jonów pochodnych, bowiem przy dużej energii jonizacji cząsteczka rozpada się na fragmenty. Przy dużej dokładności określenia masy możliwe jest odróżnienie różnych jonów o zdawałoby się tej samej masie molowej, w oparciu o różnice na trzecim czy czwartym miejscu po przecinku i dodatkowe sygnały od izotopów. Dla większego ułatwienia analizy oprogramowanie aparatów MS zwykle zawiera bibliotekę znanych widm masowych, z którą można porównać wyniki.
No dobra - mamy więc zestaw analityczny. Teraz tylko kwestia dobrania warunków i sprawdzenia czy wyniki zgadzają się z czymś nam znanym. Początkowo użyto techniki chromatografii gazowej, w której rozdział następuje w długiej kolumnie przez którą przepuszcza się gaz obojętny, a próbka jest odparowywana na małej grzałce, jednak ten sposób nie dał żadnych ciekawych wyników. Najprawdopodobniej użyta toksyna była mało lotna lub rozkładała się przy odparowaniu próbki.
Musiano więc przestawić się na technikę chromatografii cieczowej, z próbką rozpuszczaną w odpowiednim rozpuszczalniku. Jest to metoda o tyle gorsza, że do roztworu przechodzi dużo przeszkadzających substancji.
Dopiero teraz dało się wyłapać charakterystyczny sygnał, który pojawiał się we wszystkich trzech próbkach - roślinnym proszku, sosie curry i próbkach pobranych od otrutych. Widmo masowe pokazało stosunkowo dużą masę cząsteczkową, leżącą w zakresie w którym pojawia się wiele alkaloidów roślinnych. Po porównaniu masy głównego jonu i fragmentów cząsteczki z biblioteką zarejestrowanych widm nie otrzymano konkretnego wyniku - widocznie było to trucizna na tyle rzadko spotykana, że nie zapisano jej analiz. Jednak analiza zarejestrowanych fragmentów i wiedza na temat tego w jaki sposób przejonizowane cząsteczki rozpadają się w spektrometrze, pozwoliła dojść analitykom do wniosku, że musi być to któraś z toksyn z grupy akonityn.
Skrupulatne przejrzenie literatury doprowadziło badaczy do najbardziej prawdopodobnego wyniku - pseudoakonityny, toksyny z rosnącej w Indiach rośliny z rodzaju tojad, której europejski odpowiednik jest też znany jako ogrodowa roślina ozdobna. Aby potwierdzić przypuszczenia, laboratorium poprosiło ogród Kew Gardens o próbki roślin.
Tojad mocny, nazywany też mordownikiem, jak to wprost nazwa sama wskazuje jest rośliną silnie trującą. Należy do rodziny jaskrowatych, konkretnie plemienia
Delphinieae, wraz z ostróżką i ostróżeczką. Jest byliną o charakterystycznych, głęboko podzielonych liściach i ukwieconym pędzie dorastającym do półtora metra wysokości. Kwiaty duże, intensywnie granatowe z charakterystycznym "hełmem" z przerośniętego piątego płatka. Z tego też powodu nazywany był także błękitnym lub mnisim kapturem. Występuje w Europie głownie na terenach górskich i pogórzu, kilka stanowisk znanych jest z polskich Karpat. Bardziej rozpowszechniony jest tojad mołdawski, rosnący też na wyżynach i w dolinie Wisły. Pojedyncze stanowiska na nizinach to głównie wynik ucieczki z ogrodów.
Ze względu na bardzo efektowny wygląd i intensywną barwę, tojad jest uprawiany jako roślina ozdobna. Wiele osób może go mieć u siebie nie wiedząc z jak niebezpieczną rośliną mają do czynienia, wyhodowano liczne odmiany o kolorach białym czy różowym, z plamkami czy jasnym obrzeżeniem. Jest uważana za jeden z klasycznych kwiatów tradycyjnego angielskiego ogrodu.
Właściwości tojadu poznano już bardzo dawno. Wedle mitologii greckiej miał powstać w miejscu, gdzie na ziemię spadła ślina Cerbera, demonicznego psa pilnującego wejścia do Hadesu. Czarodziejka Medea miała zatruć tojadem wino przeznaczone dla Tezeusza, gdy ten zażądał należnego mu tronu. Atena użyła tej rośliby aby zamienić dumną Arachne w pająka. Przypuszcza się, że posłużyła do otrucia cesarza Klaudiusza, któremu podano zatrutą potrawę z grzybów, przy czym szybkie pojawienie się objawów i drętwienie języka nie bardzo pasowały do trujących grzybów; podobne podejrzenia dotyczą śmierci jego syna Brytanika. W późniejszych wiekach zyskała sławę "roślinnego arszeniku".
Ludy zamieszkujące Syberię, Kazachstan i Kamczatkę, oraz Ajnowie będący pierwotnymi mieszkańcami północnych wysp archipelagu japońskiego, używały soku z tojadu do zatruwania strzał i wnyków podczas polowania na niedźwiedzie czy łosie.
Podobnie jak inne silnie trujące rośliny, tojad był dawniej używany w medycynie. Skoro bowiem toksyna działa na organizm tak silnie, to w odpowiednio dobranej dozie powinna być silnie lecznicza. Medycyna europejska stosowała tojad i preparaty pochodne jako środek pobudzający czynności organów. W bardzo małych dawkach wskutek podrażnienia zakończeń nerwowych poprawiała pracę układu pokarmowego i krążenie, równocześnie uspokajając pracę serca. Wyciągi użyte zewnętrznie powodowały porażenie nerwów czuciowych a co za tym idzie lokalne znieczulenie. Używano go jednak ostrożnie, bowiem naturalny surowiec posiadał mocno zmienną zawartość toksyny i trudno było precyzyjnie dobrać dawkę.[1]
W medycynie chińskiej i ayurwedyjskiej lokalne gatunki tojadu są środkiem rozgrzewającym i zwiększającym poziom energii w nerkach, stąd też chętnie używany w przewlekłych chorobach związanych ze starością, których przyczyną miało być "wychłodzenie" i gromadzenie się "wilgoci". Roślina musiała być w tym celu odpowiednio przygotowana aby obniżyć jej toksyczność. Najczęściej brano pojawiające się jesienią bulwki korzeniowe i bardzo długo gotowano, przez co toksyna bądź częściowo rozkładała się, bądź była wymywana. Gorzkie w smaku bulwki zawierają skrobię, dlatego lokalnie są spożywane po prostu jako warzywo. Łukasz Łuczaj miał okazję spróbować takiego właściwie przyrządzonego dania.[2]
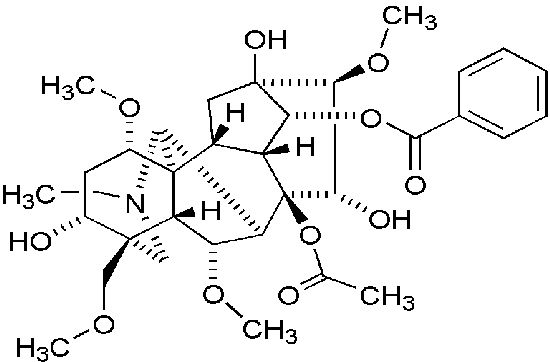
Główną toksyną europejskiego tojadu jest akonityna, alkaloid o dość skomplikowanej, przestrzennej budowie:
Należy do najsilniejszych toksyn roślinnych, dawka śmiertelna czystej substancji to zaledwie 1 mg/kg m.c. ustnie dla myszy, dla człowieka szacowane LD 50 to 3-4 mg/kg. Dawka toksyczna przy podaniu dożylnym lub przezskórnym jest jeszcze niższa. Odpowiada to spożyciu około 2 gramów rośliny lub 20-30 ml nalewki. Najbardziej toksyczna jest jesienna bulwa korzeniowa oraz nasiona. Nie znalazłem informacji czy toksyna występuje też w płatkach kwiatów.
Jest stosunkowo dobrze wchłaniana przez skórę. Podczas prac ogrodowych trzeba pamiętać aby nie pobrudzić skóry sokiem z łodyg, powoduje bowiem pieczenie a potem znieczulenie, może nawet miejscowe zdrętwienie. W 2014 roku ogrodnik pracujący w rezydencji milionera Christophera O. Thompsona stracił przytomność. Po przewiezieniu do szpitala zmarł z powodu ustania czynności narządów. Po kilku tygodniach pojawiło się podejrzenie, że mógł zatruć się tojadem, który w tym czasie kwitł. Prawdopodobnie ścinając rośliny z rabaty, pobrudził ręce dużą ilością soku, dalej toksyna bądź wchłonęła się przez skórę bądź została połknięta z jakąś kanapką. Ostatecznie nie udało się tej wersji potwierdzić, bo możliwość taką zgłoszono już po pogrzebie, gdy próbki krwi były już zutylizowane. [3]
Działanie toksyczne akonityny wynika z zaburzenia działania komórek nerwowych. Przeciętna komórka nerwowa utrzymuje we wnętrzu nierównowagę ilości jonów, wyrzucając je za pomocą kanału jonowego tzw. pompy sodowo-potasowej. Jest to baryłkowaty twór ze splecionych łańcuchów białkowych, który przechodzi na wylot przez błonę komórkową. Wyrzuca on na zewnątrz dodatnio naładowane kationy sodu, przez co na błonie powstaje niewielki ładunek ujemny. Impuls nerwowy powstaje gdy kanał jonowy otworzy się, a kationy sodu napływają do komórki. Potencjał elektryczny błony najpierw staje się zerowy (depolaryzacja) a potem na krótko dodatni. Potem kanał się zamyka i w ciągu następnych kilku mikrosekund pompa wyrzuca kationy sodu na zewnątrz, przywracając pierwotny potencjał. Powstały w ten sposób impuls zmiany potencjału rozchodzi się od neuronu do neuronu, generując przepływ informacji i umożliwiając między innymi czytanie tego tekstu.

Kanały jonowe generujące impulsy mogą być otwierane dzięki czynnikom chemicznym które łączą się z odpowiednimi receptorami białkowymi mającymi połączenie z takim kanałem. Naturalne takie substancje to neuroprzekaźniki, zaś te sztuczne to najczęściej substancje pobudzające i psychoaktywne.
Akonityna po dostaniu się do organizmu przyłącza się do białkowej podjednostki alfa kanałów jonowych i powoduje, że po otwarciu takiego kanału, jego zamknięcie następuje później niż zwykle. Skutkuje to przedłużeniem fazy depolaryzacji. Jeśli wiele takich kanałów na powierzchni komórki nerwowych zostało obsadzonych przez cząsteczki akonityny, faza depolaryzacji może trwać tak długo aż neuron przestanie wysyłać i odbierać impulsy. Jeśli zaś z kolei tych niemych neuronów będzie w organizmie odpowiednio dużo, to zatrzymanie impulsów zatrzymuje pracę narządów.
Hamowanie impulsów w nerwach obwodowych skutkuje osłabieniem pracy mięśni. Dalej pojawia się porażenie mięśnia sercowego. Działanie na mózg wywołuje brak odbioru wrażeń zmysłowych. Nieskoordynowane próby odnowienia aktywności mózgowej kończą się drgawkami, zaburzeniami oddychania i krążenia. Ostatecznie śmierć następuje po ustaniu oddechu lub pracy serca, prawdopodobnie przy zachowanej świadomości.
To zdecydowanie nie jest przyjemna śmierć.
W przypadku zatrucia pokarmowego leczenie polega na zapobiegnięciu dalszego wchłaniania trucizny, poprzez płukanie żołądka i podanie węgla aktywnego. Należy sztucznie podtrzymywać oddech a czasem też pracę serca, do czasu aż receptory nie zostaną odblokowane a toksyna usunięta. Odtrutką specyficzną jest atropina, alkaloid bielunia, która ma na komórki nerwowe działanie przeciwne.
Jak wspomniałem, tojad należy do rodziny jaskrowatych a konkretnie do "plemienia"
Delphinieae. Do tej samej grupy należy inny popularny kwiat
ostróżka, oraz częsty chwast polny
ostróżeczka. Obie te rośliny także mogą zawierać substancje o podobnej budowie, głównie metyllykakonitynę. Toksyna ostróżki ma działanie kilkukrotnie słabsze od akonityny a ponadto występuje w dużo mniejszej ilości. Ze względu na działanie na receptory nikotynowe była testowana jako środek pomagający rzucenie palenia, ma ponadto działanie przeciwstawne do cannabioidów. Wyciągi z ostróżki są czasem stosowane w "ekologicznych" opryskach przeciwko bielinkowi kapustnemu i innym owadom będącym szkodnikami warzyw.
Z kolei ostróżeczka polna, będąca chwastem, i uprawiana jako ozdobna ostróżeczka ogrodowa zawierają niewielką ilość alkaloidów, które od dawna są wykorzystywane w preparatach przeciwko wszom i pchłom.
Jak skończyła się sprawa o otrucie Cheema?
Gdy już dowiedziono, że użytą trucizną był tojad, pozostawało tylko udowodnić Lakhvir Singh winę. Znaleziono przy niej roślinny proszek będący sproszkowanym indyjskim gatunkiem tojadu
Aconite forex, zawierającym pseudoakonitynę, a więc ten sam alkaloid który ostatecznie po długich badaniach udało się wykryć w próbkach od zatrutych. Ona sama tłumaczyła jednak, że to używane przez nią zioła i że niczego byłemu narzeczonemu nie dosypywała.
Sprawdzono skąd mogła mieć proszek - kilka tygodni wcześniej była na krótko w Indiach, tam kupienie większej ilości nie nastręczało żadnych trudności. Zeznania Gurjeet, którą ostatecznie udało się odratować, także dużo wniosły. Próby definitywnego zakończenia poprzedniego związku były dość burzliwe. Singh jeszcze wielokrotnie przychodziła do domu byłego kochanka, robiąc mu awantury i strasząc, że załatwi deportację jego narzeczonej. W międzyczasie wyszło na jaw, że Cheema oszukiwał narzeczoną, mówiąc jej że rozstał się z panią Singh trzy lata temu i teraz są tylko przyjaciółmi. Była zaskoczona gdy dowiedziała się, że jeszcze niedawno spędzała u niego wolne dni. W Boże Narodzenie 2008 narzeczeni uczestniczyli w spotkaniu towarzyskim, na którym znalazła się też Singh. Gurjeet próbowała po przyjacielsku załatwić sprawę mówiąc jej, że wie o wszystko o ich wieloletnim romansie i radziła jej, że powinna jednak mimo wszystko zająć się mężem i trójką dzieci. To jednak nie poskutkowało.
Na początku stycznia 2009 Singh znów przyszła do domu kochanka, robiąc awanturę o to, że narzeczeni już ze sobą śpią choć nie mają ślubu. Wiedziała to bo zakradła się pod dom i podglądała ich przez okno. Dwa tygodnie później narzeczeni zatruli się sosem curry.
Kolejną zastanawiającą okolicznością było wcześniejsze zatrucie. Cheema zatruł się na początku grudnia potrawką warzywną i kilka dni spędził w szpitalu. Stało się to kilka dni po poważnej rozmowie, podczas której odtrącił Singh. Już wtedy podejrzewał, że dodała mu czegoś do jedzenia, ale zbagatelizował sprawę, bo w końcu zatrucie nie było silne.
Za dowód rozstrzygający uznano jednak zeznania dwóch lokatorów Cheemy, którzy po południu w dniu tragicznego zdarzenia, widzieli jak Singh przyszła do domu byłego kochanka, otwierając sobie własnymi kluczami. Nie bardzo się orientowali jak to między nimi ostatecznie było i jak się dogadali, a ponieważ widywali ją już wcześniej, nie widzieli niczego podejrzanego w tym, że skierowała się do kuchni i zaglądała do lodówki.
Na początku 2010 roku Lakhvir Singh została skazana na dożywocie z możliwością przedterminowego zwolnienia dopiero za 23 lata. O tym jak wyjątkowy i rzadki jest to przypadek świadczy to, że poprzedni wyrok w sprawie o otrucie tojadem zdarzył się w Wielkiej Brytanii w roku 1892.
---------------------------------
Źródła:
[1] http://www.independent.co.uk/news/uk/home-news/gardener-dies-after-brushing-against-deadly-wolfsbane-flower-on-millionaires-estate-9845675.html
[2] http://rozanski.li/278/tojad-mocny-aconitum-napellus-l-ranunculaceae-jak-to-dawniej-z-nim-bylo-w-medycynie/
[3] http://lukaszluczaj.pl/tojad-lek-pozywienie-i-trucizna-w-jednym/
* https://www.chemistryworld.com/feature/its-a-bloody-business/8378.article
* http://akrokorinthos.blogspot.com/2013/11/the-poison-aconite-or-wolfsbane.html
* https://en.wikipedia.org/wiki/Aconitine
* https://en.wikipedia.org/wiki/Methyllycaconitine
* http://murderpedia.org/female.S/s/singh-lakhvir.htm