Nowa odmiana alotropowa azotu o potencjale wybuchowym
Odkrycie nowej molekularnej odmiany alotropowej pospolitego pierwiastka, która nie jest po prostu faza krystaliczną o innym ułożeniu cząsteczek, to w ostatnich latach raczej rzadkie odkrycie. Tworzy się takie nowe odmiany głównie dla węgla, a dla innych niemetali większość takich dokonań miała miejsce lata temu.
Dlatego ostatnie odkrycie na temat azotu jest interesujące - stworzono cząsteczkę złożoną wyłącznie z azotu, która jest obojętna elektrycznie i nie wymaga ultra niskich temperatur aby istnieć, udało się ją w pewnych warunkach zagęścić do cieczy. Oraz jest bardzo energetyczna i ma potencjał wykorzystania w materiałach wybuchowych.
 |
Najbardziej znaną formą azotu jest cząsteczka dwuatomowa, w której atomy są połączone silnym wiązaniem potrójnym, z kolei cząsteczki oddziałują między sobą jedynie przez słabe siły van der Waalsa, dlatego pierwiastek naturalnie jest gazem o bardzo niskiej temperaturze wrzenia. Ten fakt wysokiej energii wiązania tej formy azotu i gazowej formy powoduje, że przemiany innych związków azotu do cząsteczki dwuatomowej wiążą się z wydzieleniem dużej ilości energii i dużą zmianą objętości. Na tym w dużej mierze polega działanie wielu azotowych materiałów wybuchowych; podczas eksplozji TNT z jednego mola związku powstają trzy mole dwuatomowego azotu.
 |
| Rodnik azydkowy |
Od dawna znaną formą był jon azydkowy, w którym trzy azoty są połączone liniowo, dając niestabilny anion. Jego sole z metalami ciężkimi, azydki, to materiały wybuchowe używane w spłonkach. W 1956 otrzymano jego obojętną elektrycznie odmianę trójazot, jest to wolny rodnik ze względu na niesparowany elektron. W 2003 roku doniesiono też o formie pierścieniowej, w kształcie trójkąta.
Większy liniowy analog czteroazot jest nietrwałym gazem, który w temperaturze pokojowej łatwo rozkłada się na dwie cząsteczki dwuatomowe; stabilniejsze są jego kationy. Wykroto go jeszcze w latach 50. ale mało wiadomo o jego właściwościach. W 1999 roku odkryto kation pentazyny, który daje nietrwałe sole, jednak cząsteczki obojętnej nie wyizolowano. Istnieje cykliczna forma pentazol.
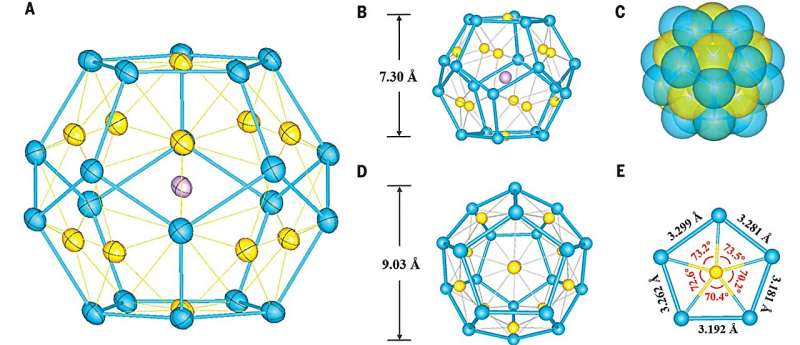
Skoro więc mamy formy zawierające 2,3,4 i 5 azotów, to czemu nie dalej? Cykliczna heksazyna pozostaje na razie hipotetyczna, ale formę liniową, sześcioazot, właśnie wykryto i wyizolowano. Można by ją opisać jako dimer rodnika triazynowego. W odmianie o największym udziale sklada się z dwóch częściu o strukturze liniowego rodnika triazynowego, połączonych wiązaniem pojedynczym i o układzie trans. Długości wiązań i geometria układu sugerują, że w uśrednionej strukturze udział ma też forma mezomeryczna z wiązaniem podwójnym pośrodku.

Synteza nowego alotropu okazała się dość prosta i łagodna - badacze przepuszczali gazowy chlor przez porowatą warstwę azydku srebra pod niskim ciśnieniem - i w temperaturze pokojowej! Sygnał nowej cząsteczki wykryto poczatkowo metodami spektroskopii IR; potem powstające gazowe produkty były zbierane na ochłodzonej powierzchni. Początkowo zimną matrycą był zestalony argon o temperaturze 10K, potem udało się też wykorzystać powierzchnię schłodzoną ciepłym azotem (77K). Sześcioazot utworzył czystą warstwę w tej temperaturze. Ponieważ istnieje bariera energetyczna przegrupowania do trzech cząsteczek N2, związek ten ma pewna stabilność.

Według obliczeń rozkład tego związku na trzy cząsteczki azotu skutkuje uwolnieniem energii 180 kcal/mol, co stanowi 2,2 razy więcej niż przy rozkładzie trotylu TNT. Oczywiście tej konkretnie cząsteczki raczej się nie zastosuje jako materiału wybuchowego, jest za bardzo reaktywna, ale może w przyszłości uda się otrzymać bardziej stabilną strukturę opartą na jej formie. Przychodzi mi też do głowy możliwość wykorzystania jako materiał pędny w warunkach kosmicznych, gdzie bez izolacji cieplnej wszystko jest dostatecznie zimne aby można było przechowywać heksanitrogen.
https://www.nature.com/articles/s41586-025-09032-9
Makeup sprzed 2700 lat
 |
| Wazonik na Kohl i patyczek do aplikowania, okres Nowego Państwa, Starożytny Egipt. Met Museum |
Na starożytnym cmentarzu Kani Kofter koło Dere Pemeyan w Iranie, w grobie z trzeciej fazy epoki żelaza, a więc sprzed około 2700 lat, znaleziona została ceramiczna fiolka, która była na tyle szczelnie zamknięta, że zachowała się jej zawartość. Czarny pigment był prawdopodobnie lokalną formą kosmetyku Kohl, znanego w starożytności w Egipcie i na Bliskim Wschodzie. Kohl służył do przyciemniania powiek i podkreślania brwi. Dziś w nawiązaniu do tej tradycji kohlem nazywa się preparaty do konturowania oka, przyciemniania powiek czy rzęs
Nakładające się analizy przy pomocy technik: spektroskopii Ramana, XRF i rentgenowskiej krystalografii proszkowej pozwoliły rozwiązać skład. Głównymi czarnymi pigmentami okazał się tlenek manganu w formie piroluzytu oraz grafit mineralny, mniejszą domieszkę stanowiły tlenki żelaza. Oprócz nich mniejszy składnik stanowiły minerały ilaste, kwarc i skaleń, być może stanowiące bazę lub będące zanieczyszczeniem z ceramicznych naczyń w których przygotowywano pigment. Nie znaleziono natomiast żadnych pozostałości po organicznych lepiszczach - niektóre składniki takich kosmetyków, jak guma arabska czy białko jaja mogły się rozłożyć, ale lipidy z tłuszczu zwierzęcego czy terpenoidy z wyciągów roślinnych powinny dać jakiś ślad. Możliwe więc, ze pigment był przechowywany jako suchy proszek a do zastosowań kosmetycznych był rozrabiany lub używano go na sucho.
Użycie jaki pigmentu grafitu jest nietypowe jak na podobne starożytne czernidła, w dodatku najwyraźniej jest to grafit mineralny a nie sadza. Częściej starożytny kohl zawierał siarczek arsenu, czarne tlenki żelaza i manganu, a jeśli już pojawiał się węgiel to w formie sadzy czy czerni kostnej. Można to jednak wyjaśnić dostępnością surowca - w rejonie Iranu, z którego pochodziła buteleczka, wydobywano grafit i używano na przykład do wykańczania powierzchni ceramiki. Archeolodzy spekulują, że mogły zadecydować też względy użytkowe; grafit daje metaliczny połysk oraz jego cząstki dobrze trzymają się skory. Tradycyjnie używane w takich formułach minerały stibnit, czyli siarczek antymonu, i galena czyli siarczek ołowiu, także mają połysk podobny do grafitu.
https://onlinelibrary.wiley.com/doi/10.1111/arcm.13097
https://archaeology.org/issues/may-june-2024/digs-discoveries/near-eastern-lip-kit/
Przypomina to zeszłoroczne odkrycie czerwonej maści, może czerwieni do powiek a może szminki, sprzed 4000 lat w irańskiej miejscowości Jiroft. Substancja znaleziona w kamiennej fiolce zawierała głównie czerwony hematyt, z dodatkiem ciemniejszych pigmentów manganitu i braunitu i domieszką anglezytu, i była zmieszana ze spoiwem z roślinnych wosków, oleju i być może jakimś materiałem roślinnym. Wypadkową tego składu powinna być ciemnoczerwona maść. Spoiwo organiczne datowano metodami radioweglowymi na zakres od 1936 do 1687 p.n.e.
https://www.nature.com/articles/s41598-024-52490-w
Elektroosadzanie złota w kwarcu
Złoto jest generalnie pierwiastkiem rzadkim i jego przeciętne stężenie w skałach skorupy ziemskiej jest niskie. Najczęściej pojawia się jako domieszka w rudach innych metali, miedzi, srebra, arsenu, czasem jako rzadkie minerały w połączeniu z tellurem. Rozproszone skupiska w postaci metalicznej zostają wyseparowane przez erozję i tworzą wtórne złoża w żwirach rzecznych, gdzie złoto zagęszcza się we frakcji ciężkich ziaren. Najbardziej jednak cenione są złoża żyłowe rodzimego złota, gdzie wydrążenie szybu wzdłuż osi żyły daje bardzo korzystny stosunek produktu do skały płonnej. Dlatego też takie złoża są poszukiwane i badane są warunki ich powstawania.
_(17161279802).jpg) |
| Złoto na kwarcu, Kalifornia |
Jednak mechanizm powodujący, że rządki pierwiastek skupia się w lite skupienia w wąskich szczelinach skalnych, nie jest do końca jasny. Złoto pojawia się wtedy w wąskim paśmie w obrębie szczeliny lub wzdłuż osi przecięcia dwóch szczelin, towarzyszy mu kwarc i piryt. Krystaliczny kwarc powstaje w wyniku osadzania krzemionki w warunkach hydrotermalnych, z przegrzanej wody z rozpuszczonymi minerałami, utrzymywanej przez ciśnienie grubo powyżej 100 stopni C w stanie płynnym. Skała musi być więc zakopana pod osadami na dużej głębokości, ma kontakt ze złożami hydrotermalnymi, które są podgrzewane przez systemy wulkaniczne, a szczeliny powstają w wyniku ruchów sejsmicznych. Problem w tym, że rozpuszczalność złota i jego związków w takich wodach podziemnych jest bardzo niska, żyły złotonośne mogą być bardzo cienkie i żeby osadzić aż tyle pierwiastka, przez wąski przekrój żyły musiały przepłynąć jakieś nieprawdopodobne ilości takiej wody.
W ostatnich latach proponowano dla takich sytuacji alternatywne rozwiązania, jak powstawanie nanoczątek złota, które omijają ograniczenia rozpuszczalności, i agregują się w żyłach na zasadzie przemiany zol/żel. Teraz pojawiła się bardzo ciekawa propozycja, poparta eksperymentem, która może się okazać słuszna.
O kryształach kwarcu wiadomo od dawna, że jest piezoelektryczny - odkształcenie mechaniczne kryształu generuje ładunki elektryczne, nieraz na tyle duże, że powodujące przeskok iskry. Wykorzystuje się to od dawna choćby w zapalniczkach gazowych. Skoro więc w żyle powstają kryształy kwarcu, w wyniku ruchów tektonicznych są one poddawane naprężeniom, i są zanurzone w roztworze zawierającym sole metali, to może złoto wydziela się poprzez znany od dawna proces elektroosadzania?
W ramach testu zanurzono kryształy kwarcu w roztworze soli złota (w teście laboratoryjnym był to najwyraźniej chlorozłocian) i poddano je naciskowi. Po upływie dłuższego czasu obejrzano kryształy pod mikroskopem. Wytrąciło się złoto! Powstały krystaliczne nanocząstki złota, miejscami warstwy pokrywające powierzchnię. Nowe porcje złota chętniej dołączały się do już utworzonych wytrąceń, które działały jak elektroda i zbierały z powierzchni ładunek. Tym sposobem narastanie bryłek jest dużo szybsze niż powolne, samoistne wytrącanie przy przekroczeniu granicy rozpuszczalności.
Przy czym ten proces wcale się nie wyklucza z wcześniejszą propozycją, może on tworzyć nanocząstki złota, które potem przyłączają się do agregatów.
https://www.nature.com/articles/s41561-024-01514-1#citeas