Mając chwilkę czasu w laboratorium, zabawiłem się w rozdzielanie na składniki czarnych markerów, jakie były na stanie pracowni do szkła i plastiku:

O chromatografii kiedyś już pisałem (artykuł). Jest to technika rozdzielająca mieszaniny na poszczególne składniki, pozwalająca dzięki porównaniu ze wzorcami też na ich oznaczenie. Odkryta na początku XX wieku przez rosyjskiego botanika Cwieta stała się dziś jedną z podstawowych technik analitycznych.
Cały proces opiera się o zachodzenie dwóch przeciwstawnych zjawisk - adsorpcji substancji na powierzchni chłonnego materiału i jej wypieraniu przez cząsteczki rozpuszczalnika. To na ile mocno substancja zwiąże się z podłożem zależy w dużej mierze od tego co to jest za substancja i jakie jest to podłoże.
Na adsorbencie będącym materiałem polarnym, wchłaniającym wodę, łatwiej będą się osadzać substancje polarne, hydrofilowe, zaś aby je dobrze wymyć trzeba użyć także odpowiednio silnego, polarnego rozpuszczalnika. Podobne do podobnego. Siła oddziaływania substancji z podłożem zależy od budowy i wielkości cząsteczki - obecność atomów niemetali z wolnymi parami elektronowymi (tlen, siarka, azot) sprzyja tworzeniu wiązań wodorowych, które mocniej wiążą cząsteczkę. Dla układów gdy podłoże jest niepolarne, tłuste, siłę wiązania zwiększają grupy węglowodorowe. Duża cząsteczka niepolarna może się lepiej wiązać z niepolarnym podłożem niż mała.
Natomiast siła z jaką rozpuszczalnik wymywa substancję zależy od tego jak silnie z nią oddziałuje i od tego na ile silnie wiąże się z podłożem.
Wszystkie te efekty powodują, że różne substancje mają różną siłę osadzania się na materiale chłonnym, czyli różne powinowactwo. Jeśli umieścimy mieszaninę na początku masy adsorbenta i będziemy przepuszczać przez niego rozpuszczalnik, składniki najsłabiej oddziałujące z podłożem popłyną najszybciej, a te najmocniej popłyną najwolniej. Przypomina to sytuację gdy na stadionie sportowym do biegu na kilometr zgłosi się mieszanka młodzików i dobrze wytrenowanych sportowców - ci lepsi szybko oddzielą się od słabszych, tworząc osobną grupkę.
Spróbujmy zrozumieć na czym polega to zróżnicowanie prędkości. Powierzchnia ziaren podłoża jest na tyle duża, że rozpuszczona porcja substancji nie przepływa po prostu kanalikami, tylko zostaje cała skutecznie wyłapana i osadzona. Ale zarazem z tyłu czysty rozpuszczalnik wymywa substancję i przeprowadza przez nasycone ziarna do przodu, gdzie osadza się na jeszcze nie pokrytym podłożu. Bardziej więc przypomina to ruch wydmy gnanej wiatrem niż prosty przepływ. Jeśli substancja lepiej oddziałuje z podłożem, jest słabiej wymywana przez rozpuszczalnik. W efekcie więcej czasu pozostaje związana i zostaje w tyle za lepiej wymywanymi.
W ten sposób skomplikowane mieszaniny kilkunastu czy kilkudziesięciu składników mogą zostać rozdzielone.
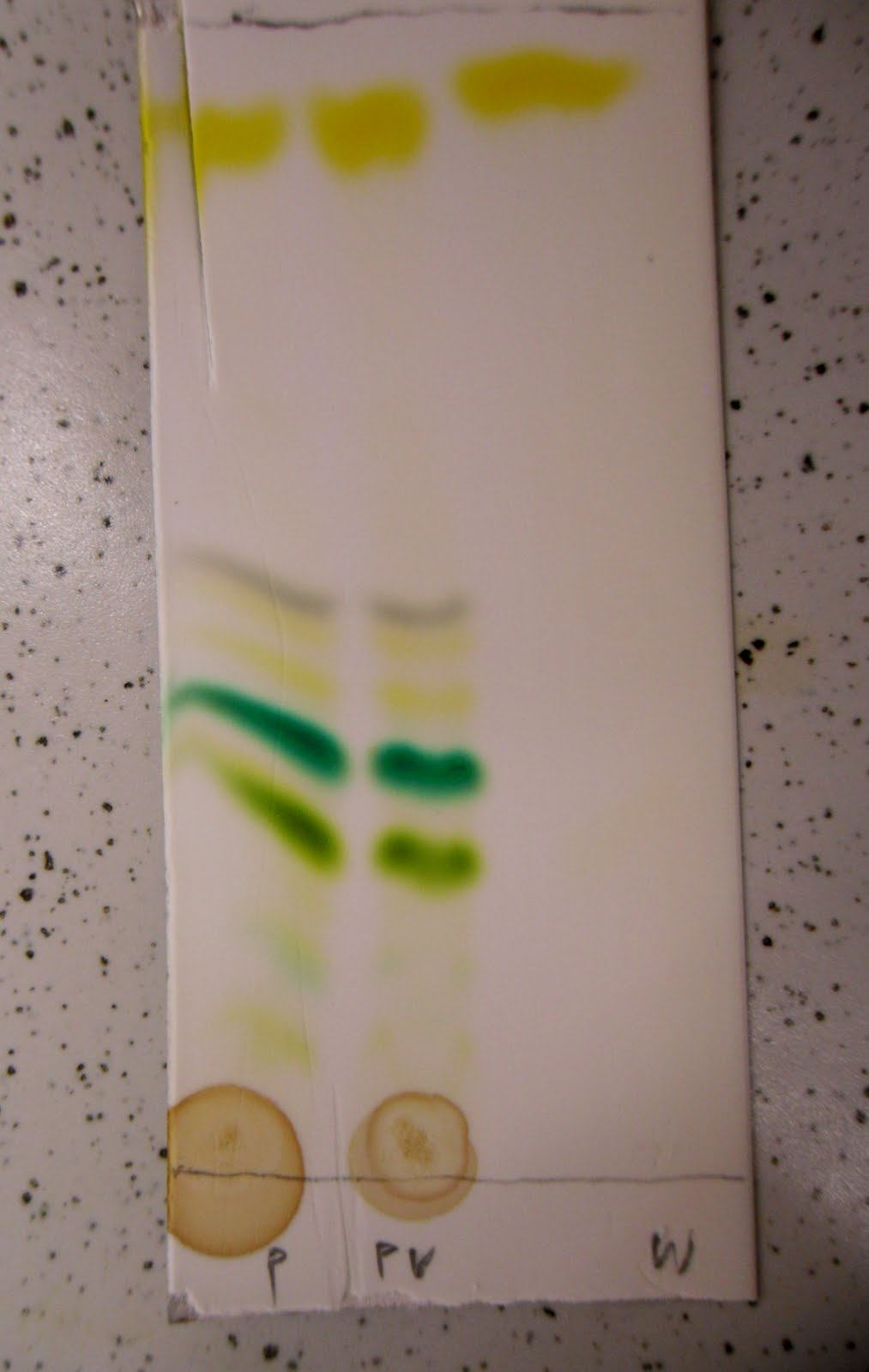 |
| Barwne składniki wyciągu z zielonych liści |
W moim przypadku podłożem, adsorbentem, była cienka warstwa masy krzemionkowej osadzona na aluminiowej folii. Miałem do użytku na pracowni cały arkusz, który zużywałem przy kolejnych syntezach podczas sprawdzania, czy reakcja zaszła, a gdy został mi na koniec taki nierówno wycięty kawałek, postanowiłem użyć go do opisanego tu doświadczonka.
Na starcie, nad brzegiem płytki, naniosłem kropki czterema czarnymi markerami, jakie akurat miałem dostępne. Jako naczynia użyłem najmniejszej zleweczki i przykrywki od naczynka pomiarowego. Nie pamiętam jaki dokładnie był skład rozpuszczalnika, ale generalnie był to chlorek metylenu z odrobiną octanu etylu, bo tego akurat używałem.
Aby proces chromatograficzny zachodził, należało wytworzyć ruch rozpuszczalnika w materiale płytki, użyłem tu znanego zjawiska podciągania kapilarnego - wlałem do zlewki taką ilość rozpuszczalnika, aby cały dolny brzeg był zanurzony, ale też aby zarazem same plamki mi się w nim nie moczyły, i przykryłem całość nakrywką, aby nie parowało. Zanim płytka nasiąknęła co górnego brzegu minęło kilka minut, toteż film nakręcony podczas procesu trochę przyspieszyłem:
Jak widzicie cztery z pozoru identycznie czarne plamki rozwinęły się w różnokolorowe pasma.
Generalnie rzecz biorąc nie ma czarnych barwników. Czerń powstaje wtedy, gdy substancja pochłania tak dużo światła, że oko nie rejestruje konkretnego koloru. Zwykle jednak po mocnym rozjaśnieniu czerń okazuje się być bardzo, bardzo ciemnym konkretnym kolorem. Mogą istnieć czarne pigmenty, to jest stałe substancje pochłaniające w dużym stopniu wszystkie kolory światła, tu najczęściej używany jest węgiel. Trudno jednak zastosować pigment w farbach wodnych i w flamastrach, w których tusz z wkładu przesiąka do końcówki przez porowaty materiał, działający raczej jak sito dla stałych cząstek.
Producenci używają więc mieszanek różnych barwników o dużej sile barwienia. Gdy na dany barwnik pada światło białe, pochłania on z zakresu pewne kolory a odbija inne. Jeśli dobierzemy barwniki tak, że każdy kolor będzie po trochę pochłaniany, mieszanka będzie wyglądała na czarną. A jak pokazało moje małe doświadczenie, różni producenci lubią też używać różnych, unikalnych mieszanek:

W przypadku czwartego markera, Pentel Pen, składniki okazały się w układzie na tyle dobrze rozpuszczalne, że bez wyraźnego oddzielenia popłynęły na sam koniec, tworząc czarną plamkę.
Ten obraz poszerzyć może badanie wyglądu płytki w ultrafiolecie, ujawniające składniki nie widoczne gołym okiem. Substancje fluoryzujące świecą własnym światłem:

Po co niewidoczny gołym okiem składnik w markerach? Ponieważ świeci w ultrafiolecie, to musi go też pochłaniać, jest to więc zapewne składnik chroniący pozostałe barwniki przed degradacją na świetle, powstrzymujący blaknięcie rysunków.
Różnice w składzie tuszu markerów, ale też tuszu długopisów czy atramentu piór wiecznych mają istotne znaczenie w kryminalistyce, aby wyryć czy badane dokumenty, na przykład testament, nie były później uzupełniane. Jeśli sprawca użył innego długopisu, różny skład potwierdzi dopiski. Oczywiście nie wkładamy w tym celu dokumentu do naczynia z rozpuszczalnikiem aby spojrzeć na powstające kolorowe plamki. Bądź pobiera się drobną próbkę z dokumentu i bada którąś do dokładnych technik chromatograficznych, jak wysokosprawna cieczowa, bądź wyznacza technikami nieinwazyjnymi, jak spektroskopia Ramana czy UV-Vis
A jak wykonać podobne doświadczenie u siebie w domu? Specjalistycznych płytek TLC nie trzeba kupować. Za cienki materiał chłonny wystarczy arkusz grubej bibuły, na przykład gęsty filtr do kawy, można też próbować ze sztywnym, kredowym papierem. Mi kiedyś udało się to z papierem do kserowania.
Wycinamy z papieru pasek o takiej szerokości aby zmieściły się nam kropki wszystkich flamastrów jakie chcemy zbadać, długi na kilka centymetrów. Znajdujemy wysokie naczynie o płaskim dnie, może to być słoik, szklanka, opakowanie po czymś, tak aby nasz pasek się w nim mieścił.
Teraz kwestia rozpuszczalnika - dość dobrymi, mocno wymywającymi, jest spirytus i zmywacz do lakieru do paznokci. Jeśli okażą się zbyt mocne i podczas próby wszystkie kolory od razu pójdą do góry, możemy spróbować domieszać jakiegoś słabszego składnika, może to być na przykład jakiś rozpuszczalnik do usuwania tłustych plam. Jeśli badamy markery nierozpuszczalne, pomocne może być dodanie odrobiny wody - wprawdzie jest bardzo polarna, ale gdy składniki barwne się w wodzie słabo rozpuszczają, woda może pogorszyć ich wymywanie z papieru i spowolnić. Tu już trzeba sobie poeksperymentować.

Przygotowaną mieszankę wlewamy na dno naszego naczynia, wkładamy pasek papieru z naniesionymi u dołu kropkami markerów tak aby opierał się o ściankę. Ponieważ nasiąkający papier traci sztywność, aby się nam nie przewrócił i nie wpadł możemy bądź zawinąć górny brzeg na brzegu naczynia, lub użyć spinacza do papieru, ewentualnie przewlec nitkę przez otwór w papierze i podwiązać. Pasek nie powinien przylegać do ścianki naczynia, rozpuszczalnik będzie wówczas podsiąkał w szczelinie między nimi i całość się rozmyje. Naczynie czymś przykrywamy aby rozpuszczalnik nie parował i czekamy aż cały pasek nasiąknie.




 .
.
















