Ostatnie doniesienie astronomów na temat odkryć dotyczących planety Wenus ma w sobie tyle chemicznych wątków, że warto się w nie zagłębić.
Wprawdzie szczegóły procesu powstania życia nie są dla nas jeszcze jasne, ale wydaje się mało prawdopodobne, aby zaszedł on tylko raz, na jednej skalistej planecie. Toteż nie jest wcale naukowym dziwactwem szukanie śladów życia w kosmosie. Tu jednak pojawia się problem - jak, poza bardzo oczywistymi sytuacjami, w rodzaju sfery Dysona, kierowanych związek radiowych czy modulowanego strumienia neutrin, mielibyśmy wykryć, że na planecie, na jaką nie możemy osobiście polecieć, dzieje się coś poza geologią? Że istnieje tam jakieś życie?

Z większych odległości możemy sobie na planetę tylko popatrzeć bez szczegółów, więc póki jej mieszkańcy nie osiągną takiego stanu zaawansowania, że cywilizację będzie widać, to jedyne co nam pozostaje, to wykrywanie subtelnych sygnałów modyfikacji stanu planety. Organizmy żywe muszą w jakiś sposób generować energię aby produkować złożone molekuły budulcowe. Muszą więc gromadzić związki poddające się reakcjom redukcji lub utlenienia, do ich przemian z pewnością będą wymagały pewnych katalizatorów, a tymi najczęściej są związki rzadkich pierwiastków. Metabolizm tych organizmów spowoduje zmniejszenie ilości substratu energetycznego, oraz wyprodukowanie substancji odpadowych. Są ziemskie bakterie, które "oddychają" utlenionymi związkami żelaza, zamieniając je w zredukowane. Inne redukują azotany do amoniaku, siarczany do siarki, utleniają metan, redukują arszenik... A pewna szeroka grupa organizmów przetwarza energię świetlną i przy jej pomocy zamienia dwutlenek węgla i wodę w związki organiczne i odpadowy tlen.
Wszystko to powoduje, że organizmy zasiedlające globalnie całą planetę zaczną z czasem zauważalnie zmieniać jej stan. Stąd powstał pomysł poszukiwania na planetach związków sygnałowych - trudnych do wytworzenia przez procesy geologiczne, mogących być końcowymi produktami metabolizmu organizmów, niezależnie od tego na jakiej chemii są oparte. Zwłaszcza takich, jakie powstają przy udziale jedynego życia, jakie na razie znamy - tego ziemskiego. Jeśli zbadamy planetę wykazującą pewne podobieństwa do naszej, to mamy spore szanse, że występujące tam życie będzie miało dużo wspólnych elementów. Może nie taki sam nośnik genetyczny, może nie alfa-aminokwasy, ale raczej metabolizm obracający głównie związkami węgla, azotu, tlenu, siarki i fosforu.
Wśród kilku wytypowanych takich związków sygnałowych, obok metanu, tlenosiarczku węgla i innych, pojawiła się także fosfina, czyli fosforowodór. A to ze względu na kilka właściwości utrudniających spontaniczne powstawanie na planetach typu ziemskiego. No i teraz właśnie tę fosfinę wykryto na Wenus - gorętszej "siostrze" Ziemi. Co w świetle powyższych rozważań ma bardzo ciekawe implikacje.
Fosfina
Właściwie jest to fosforowodór, będący analogiem bardziej pospolitego amoniaku. Czysty związek PH3 to bezbarwny gaz, nie posiadający wyczuwalnego zapachu. Łatwo łączy się sam ze sobą w dimer P2H6, który już ma brunatny kolor i nieprzyjemny zapach stęchlizny lub czosnku, w mniejszych ilościach pachnie jak karbid. Jeśli ktoś używał karbidu do odstraszania kretów lub do strzelania z puszek, musi kojarzyć charakterystyczny smrodek. Karbid przemysłowo otrzymywany jest przez prażenie wapienia z węglem, powstały węglik reaguje z wodą dając bezwonny acetylen. Ślady fosforanów w wapieniu powodują powstanie domieszki fosforku wapnia, która w reakcji z wodą daje fosfinę i jej dimer. Ponieważ tak powstały gaz jest potem zużywany do spawania, acetylen techniczny także nabiera tego specyficznego zapachu, zaś linie widmowe fosfiny są zauważalne w widmie światła palnika.
Sam w sobie fosforowodór jest związkiem silnie trującym, co najmniej tak bardzo jak cyjanowodór. Z tego powodu fosforek glinu używany jest czasem jako środek do trucia szczurów i insektów. Spożyty przez zwierzę hydrolizuje w żołądku z wydzieleniem fosfiny; jest stosowany zwłaszcza wtedy, gdy w okolicy żyją gryzonie odporne na standardowe trutki. Może być też fumigantem do zagazowania szkodników w pomieszczeniach, zwłaszcza w przestrzeniach o trudnym dostępie, w magazynach ziarna. W Indiach szeroko stosowany jako bardzo tani środek co roku doprowadza do wielu przypadkowych lub celowych zgonów.
Wenus
Wenus to najbliższa nam inna planeta w układzie słonecznym, przy tym wykazująca dużo podobieństw. Ma podobną średnicę i masę co Ziemia, jest okryta atmosferą. Czasem bywa nazywana "gorącą siostrą Ziemi". Ostatecznie jednak jej ewolucja przebiegła inaczej. Kiedyś, gdy słońce świeciło z mniejszą siłą, znajdowała się w ekosferze, prawdopodobnie występowały na niej powierzchniowe oceany. W miarę upływu czasu rosła jednak intensywność promieniowania Słońca, co wraz z mniejszą niż Ziemia odległością spowodowało, że na Wenus zaczęło się robić gorąco. Woda powierzchniowa wyparowała. Z powodu słabego pola magnetycznego górne warstwy atmosfery nie były chronione przed wiatrem słonecznym i naładowanymi cząstkami z kosmosu.
Docierająca tutaj woda była rozbijana na składowe a bardzo leciutki wodór łatwo wyparowywał w przestrzeń kosmiczną. W efekcie Wenus stała się bardzo sucha. Brak ruchu płyt kontynentalnych i bardzo słabego, z powodu braku oceanów, wiązania w minerałach związków węgla, ten uwolniony przez wulkany gromadził się stopniowo i dziś stanowi główny składnik atmosfery (95%). Jego masa molowa jest większa niż średnia masa ziemskiej atmosfery. Przy dość podobnej grawitacji atmosfera złożona głownie z wyraźnie cięższego gazu wywołuje na powierzchni ciśnienie 90 razy większe niż standardowe na Ziemi.
Ponieważ dwutlenek węgla jest gazem cieplarnianym, to przy tak dużym stężeniu i przy mniejszej odległości od Śłońca wywołał on nagromadzenie dużych ilości ciepła. Z czasem temperatura na powierzchni zaczęła powodować rozkład niektórych minerałów i uwolnienie bardziej lotnych związków, w tym tlenków siarki. Obecnie na wysokości 40-60 km nad powierzchnią rozciąga się warstwa chmur złożonych z kwasu siarkowego i innych kwasów, która zasłania grunt, przez co w świetle widzialnym nie widać żadnych szczegółów.
 |
| Chmury na Wenus, zdjęcie z sondy Pioneer 1 |
Mniej znaczące składniki to kwas solny, kwas fluorowodorowy, tlenek węgla, ślady pary wodnej, siarkowodoru. Nie jest to więc za bardzo przyjazne środowisko. Powyżej warstwy chmur fotoliza powoduje pojawienie się tlenu. Im bliżej powierzchni, tym goręcej. Na średniej wysokości temperatura wynosi około 460 stopni C, wystarczającej do stopienia ołowiu. Przy ciśnieniu około 92 atmosfer dwutlenek węgla znajduje się blisko stanu nadkrytycznego, przypomina więc bardziej lekką, nielepką ciecz, która dobrze przewodzi ciepło.
Ciężko w takich warunkach o życie podobne do naszego. Spekuluje się jednak o możliwości przetrwania ekstremofilnych organizmów głębiej w skorupie lądowej. Inna interesująca możliwość, to przetrwanie organizmów na dużych wysokościach. Na wysokości wierzchołków chmur ciśnienie atmosferyczne jest zbliżone do ziemskiego a temperatura bliska 20-30 stopni. Ze względu na rozkład dwutlenku węgla pod wpływem ultrafioletu w atmosferze pojawia się tlen. Potencjalnie więc drobne, lekkie organizmy mogłyby utrzymywać się w tej cienkiej warstwie, gromadząc w centrach komórek konwekcyjnych. Strefa ta byłaby zresztą możliwym miejscem osadzenia latającej jak sterowiec stacji badawczej, ale to na razie pieśń przyszłości.
Artykuł
Ostatni artykuł [1] z Nature Astronomy opisuje prace, których założeniem od początku było wykrycie, że fosfina jest obecna w atmosferze Wenus. Obserwacje prowadzono naziemnie, za pomocą teleskopu Clerka-Maxwella, potem jeszcze raz przez teleskopy ALMA, mierząc widmo światła odbitego przez atmosferę Wenus w zakresie mikrofalowym. Poszukiwano tutaj charakterystycznej linii absorpcyjnej - jeśli gaz jest obecny w atmosferze, to powinien on pochłaniać ze światła pewien wycinek przy długości fali 1,123 mm. Zatem na wykresie intensywności światła przy różnej długości fali powinien pojawić się dołeczek. I tutaj pojawia się pierwszy problem - ten dołeczek nie będzie za bardzo głęboki, czyli mało będzie się różnił od szumu tła. Po drugie zaś powierzchnia odbijająca to nie ładna, równiutka skała, tylko pofalowana, zachmurzona atmosfera obracającej się planety. Efekt Dopplera powoduje, że chmury poruszające się trochę do nas lub trochę od nas będą dawały sygnał nieco przesunięty; dodatkowo sama temperatura atmosfery poszerza sygnały. Dodajmy do tego niejednorodny skład chmur i zakres fal, w którym pochłaniają różne związki, a otrzymamy sygnał nieprawdopodobnie zaszumiony. To jak poszukiwanie odcisku palca na czymś przypominającym rzeszoto.
Autorzy zastosowali odszumienie oparte o modelowanie rozkładu prędkości radialnych chmur. Otrzymali z grubsza wygładzony sygnał, w którym we właściwym miejscu pojawiał się wyraźny dołeczek. Wyglądało na to, że mają poszukiwany związek. Aby jednak skalibrować swoją metodę i z czymś ją porównać, w ten sam sposób przetworzono sygnał dla nieodległej w widmie linii absorpcyjnej półciężkiej wody HDO2. Z badań wcześniejszych sond wiemy, że na Wenus sporo wodoru zawiera cięższy izotop deuter i ile go jest, a cząsteczka zawierająca jeden prot i jeden deuter wywołuje pewne charakterystyczne dołeczki w świetle przechodzącym. Ponieważ z użyciem tego samego modelowania znaleziono sygnał związku, który na pewno jest obecny w atmosferze, wszystko wskazywało na to, że dobrze dobrano parametry i otrzymany wynik dla fosfiny to sygnał rzeczywisty a nie artefakt przetwarzania szumu.
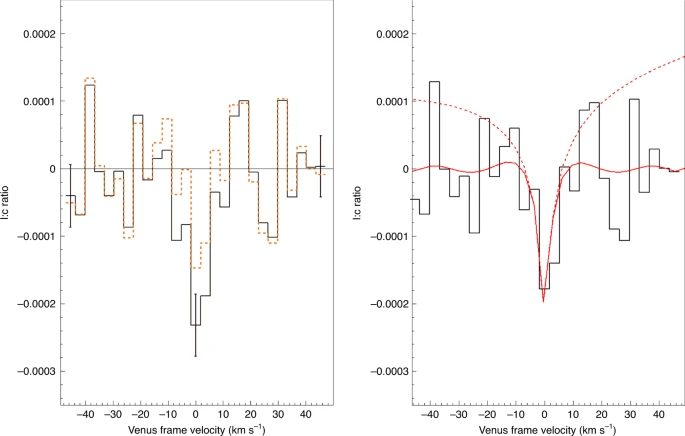 |
| Jane S. Greaves et al. |
Należało jednak wykluczyć możliwość, że przypadkiem w tym miejscu pojawiło się pochłanianie światła przez coś innego. Z wyliczeń wynikało, że obecność dwutlenku siarki (SO2) w chmurach poruszających się w naszą stronę z prędkością +2,5 km/s mogłaby wskutek przesunięcia dopplerowskiego dać sygnał w tym samym miejscu. Ponieważ jednak znamy zawartość tlenków siarki w atmosferze planety, oraz znamy intensywność pochłaniania, to wyliczono, że ten hipotetyczny dołeczek byłby zbyt mały aby wyjaśnić cały sygnał. Skoro więc tak, to pozostawała tylko jedna możliwość - to jest na prawdę sygnał fosfiny.
Ta cześć badań wygląda porządnie. Brak mi znajomości aparatu matematycznego na takim poziomie, aby coś tu ocenić. Górnicy danych może by coś na ten temat powiedzieli, ale ja nie mam kompetencji.
Fosfina to dość mocno zredukowana forma fosforu. Występuje on w tym związku na stopniu utlenienia -3. Aby sprowadzić go do takiej formy trzeba więc zwykle mocno redukujących warunków. Wykryto go już na Jowiszu i Saturnie, gdzie może powstawać w obecności wolnego wodoru, na dużej głębokości w atmosferze, gdzie panuje wysokie ciśnienie i temperatura. Sam proces redukcji i syntezy właściwego związku zużywa dużo ciepła. Wielkie planety gazowe posiadają takie warunki a ich atmosfera jest mocno redukująca.
Na Wenus jest to trochę
kłopotliwe, bo atmosfera zawiera związki tlenowe - głównie dwutlenek
węgla, do tego sporo tlenków siarki, kwas siarkowy; natomiast bardzo niewiele jest
związków wodoru, który mógłby jakoś pośredniczyć w reakcjach redukcji.
Dodatkowo związek ten jest dość reaktywny może reagować ze związkami tlenowymi i
zamieniać się w utlenione formy fosforu. Skoro więc jest obecny w
atmosferze, to jakiś proces musi go stale uzupełniać.
Autorzy przeliczają wiele możliwych dróg tworzenia fosfiny i
stwierdzają, że nie są one wystarczające aby powstawało jej stale aż
tyle. Sprawdzono takie ewentualności jak powstawanie w wyniku działania piorunów w burzach w atmosferze planety (wynik - aktywność burzowa jest zbyt niska), powstawanie z rozkładu kwasu fosfonowego produkowanego z fosforowego V pod wpływem rodników wodorkowych powstających przez fotolityczny rozkład śladów wody. Także ewentualność, że fosfina jest wyrzucana przez wenusjańskie wulkany nie bardzo pasuje do informacji o niezbyt wyraźnej aktywności geologicznej planety.
Z drugiej strony autorzy stwierdzają otwarcie, że policzenie wszystkich reakcji dla ustalenia jaka jest równowaga między formami fosforu jest trudne, bo nie wszystkie możliwe warianty zostały dobrze opisane. "Brak jest danych kinetycznych form fosforu". W części obliczeń posłużono się więc szacunkami na podstawie dużo lepiej przebadanych reakcji związków azotu. Może się więc okazać, że obfitość fosfiny w atmosferze Wenus ma źródło w jakiejś słabo znanej reakcji.
Alternatywy
Tutaj jednak ja mogę dorzucić nieco wątpliwości i alternatyw.
Najważniejszym punktem wartym zbadania są szacunki zawartości kwasu fosfonowego w atmosferze. Jest to nieco niżej utleniony kwas fosforowy, znany z tego, że w temperaturze około 200 stopni rozkłada się i dysproporcjonuje z wydzieleniem między innymi fosfiny.
4 H3PO3 → 3 H3PO4 + PH3
Autorzy przeprowadzają następujący ciąg rozważań - kwas fosfonowy nie jest stabilny w fazie gazowej. Musi być więc składnikiem kropelek zawierających kwas fosforowy i w takiej formie trwać w obiegu form fosforu. Założono, że związek powstaje wskutek reakcji rodników wodorkowych, powstałych w wyniku fotolizy śladowej ilości wody, z kwasem fosforowym. Stężenie rodników w górnych warstwach atmosfery jest niewielkie, szybko są zużywane na konkurencyjne reakcje. Na podstawie warunków w atmosferze i szybkości reakcji redukcji wyliczono, że może współistnieć w kropelce z kwasem fosforowym w ilości 6,1^-17 mol, co przy pewnej wyliczanej objętości materiału aerozolu daje 44 mg kwasu w atmosferze. Bieżący rozkład tej ilości nie daje takiego stężenia produktów jak zmierzone. [2]
Pojawia się więc pytanie, czy są inne niż tutaj założone źródła kwasu fosfonowego? W pracy z
1989 roku[3] na podstawie danych z sondy Vega2 stwierdzono, że w niższych,
gorętszych warstwach atmosfery fosfor występuje między innymi w formie
gazowego trójtlenku P4O6, mającego stanowić główny nośnik fosforu między
innymi formami. Istnienie takiej formy fosforu ma być wytłumaczeniem
powstawania mgły w dolnych warstwach atmosfery, gdzie temperatury stają
się już bardzo wysokie i mgła kwasu siarkowego powinna wyparować. Jest to o tyle istotne, że tlenek ten reaguje z
wodą tworząc kwas fosfonowy. Wody w atmosferze Wenus jest niewiele, ale może to już wystarczać. Krasnopolski szacował, że na wysokości 25
km trójtlenek fosforu stanowi 2 ppm objętości, jest go więc tam sporo.
Można więc przedstawić alternatywną drogę powstawania kwasu fosfonowego,
co uzasadnia wyższe stężenia w kropelkach kwasu fosforowego i być może
umożliwiałoby to powstawanie fosfiny w dostatecznie dużym stężeniu.
Autorzy artykułu, który jest tematem niniejszego wpisu, znają hipotezy Krasnopolskiego, bo nawet cytują tę właśnie pracę (przypis 32), ale w kontekście słabo poznanego chemizmu fosforu na Wenus, hipotetyzowanego z danych o aerozolach i ilości pierwiastka. Potem przy omówieniu potencjalnych mechanizmów powstawania fosfonianów nie odnoszą się do tego i w sumie nie wiadomo czemu - wykluczają transport tlenku, jego uwodnienie i przeniesienie do wysokości, w której panują warunki odpowiednie do reakcji? Ktoś inny w późniejszych latach obalił przypuszczenia co do istnienia tam trójtlenku? Nie znam literatury na temat chemizmu Wenus na tyle aby to ocenić, ale zupełny brak odniesienia do tej alternatywy jest zastanawiający.
A może jednak zachodzi tu całkiem zwyczajna redukcja fosforanów? Bez rodników, wyładowań i promieniowania. W atmosferze Wenus występuje pewna ilość pośredników redoks, nie zawierających wodoru i dlatego chyba nie branych tu pod uwagę. Najpospolitszy wydaje się chlorek żelaza zarówno II jak i III, który był wykrywany w chmurach na wysokości 60 km. Mechanizm powstawania mniej utlenionej formy i transportu na takie wysokości jest niejasny, możliwe jest powstawanie chlorku żelaza II w wyniku reakcji cząstek magnetytu z kwasem solnym i tlenkiem węgla.[4] Ponieważ sygnały spektroskopowe w chmurach wskazują na obecność pokaźnych ilości chlorku żelaza III [5] można rozważyć też takie drogi redukcji, jak reakcja z siarkowodorem czy dwutlenkiem siarki.
Jest to ciekawe bo nie tak dawno podczas badań, które sprawdzały, w jaki sposób mogło dojść do uwolnienia fosforu z osadów w okresie Archeanu stwierdzono, że fosforan może w stosunkowo niskich temperaturach i atmosferze beztlenowej reagować ze związkami żelaza II i redukować się do jonu fosfonowego.[6] A ten rozkłada się do fosfiny w zbliżonej temperaturze.
 |
| Ogólny schemat powstawania fosfiny w hipotezie żelazowej |
Na stronie 13 suplementów wspomniane są związki żelaza II jako brane pod uwagę wśród reduktorów, ale nie mogę się dokopać do informacji jaką konkretnie reakcję brano pod uwagę.
Czy te dwie alternatywy wystarczą do wyjaśnienia tej ilości fosfiny w sposób niebiologiczny? Przypuszczać można sobie do woli, ale może ktoś to kiedyś wysymuluje i przeliczy. Ponieważ doniesienie jest bardzo ciekawe, to zapewne inni chemicy ryją teraz po literaturze i szukają czegoś podobnego, więc za kilka miesięcy będą już jakieś analizy.
Gdyby jednak okazało się, że obecności fosfiny w tak dużych ilościach nie da się wyjaśnić reakcjami samorzutnymi, to byłby to impuls do dogłębnego zbadania tej planety. Bo odkrycie jakiegoś życia poza Ziemią, choćby mikroskopijnego, byłoby czymś wielkim, co zmieni nasze postrzeganie Wszechświata.
Errata
Dopiero po wrzuceniu artykułu udało mi się znaleźć pełne opracowanie na temat wszystkich analizowanych w publikacji reakcji. Okazało się, że postanowili wydać je osobno i na razie czeka na publikację w repozytorium. [7] Wśród rozważanych reakcji jest też redukcja żelazem II. Przy czym ewentualność bezpośredniej redukcji kwasu fosforowego żelazem została wykluczona, bowiem w dotychczasowych opisach taką redukcję przeprowadzano w wodzie, a wody jest na Wenus bardzo mało. Wciąż jednak jak na mój gust możliwa jest wersja redukcji w roztworze w kropelkach kwasu siarkowego, a więc w środowisku o wysokiej aktywności protonów. Czy zaś reakcje te są w stanie zajść w takich warunkach, chyba nie wiadomo. Znów wychodzi na jaw, że pewnych reakcji jeszcze nie zbadaliśmy dostatecznie.
------
* https://pl.wikipedia.org/wiki/Atmosfera_Wenus
[1] https://www.nature.com/articles/s41550-020-1174-4
[2] https://static-content.springer.com/esm/art%3A10.1038%2Fs41550-020-1174-4/MediaObjects/41550_2020_1174_MOESM1_ESM.pdf
[3] https://www.sciencedirect.com/science/article/abs/pii/0019103589901681
[4] Godfrey T. Sill, "Geochemical problems in the production of the Venus clouds", rozdział w: zbiorze International Astromonical Union, Symposium 40 "Planetary Atmospheres" 1969
[5] https://www.sciencedirect.com/science/article/abs/pii/S0019103516306509
[6] https://www.nature.com/articles/s41467-018-03835-3
[7] https://arxiv.org/ftp/arxiv/papers/2009/2009.06499.pdf