
Prostsza degradacja trwałych zanieczyszczeń
Zanieczyszczenia chemiczne odporne na degradację zwracają w ostatnich latach coraz większą uwagę. Niektóre substancje krążą w biosferze od dawna i jeszcze będą wypływać przez lata. W osadach rzecznych wielu krajów nadal tkwią chlorowane bifenole, wycofane w większości w latach 80. po wykazaniu ich toksyczności i wracają na powierzchnię podczas większych powodzi.
Jedną taką grupą trwałych zanieczyszczeń są związki perfluorowane, to jest z wszystkimi atomami wodoru zamienionymi na fluor. Fluorowanie węglowodory są stosowane w powłokach wodoodpornych, farbach, materiałach nieprzemakalnych, opakowaniach żywności i materiałach izolacyjnych. Jeden z nich PFOA jest stosowany przy produkcji teflonu i stanowi pozostałość w produktach z teflonem nie przetwarzanym termicznie (przy produkcji patelni jest zgrzewany i zwykle w powłoce jest tego zanieczyszczenia mało) - ale trend jaki widzę w artykułach, aby wszystkie uwolnienia substancji perfluorowanych utożsamiać z produkcją teflonu przez jedną firmę, to nadmierne uproszczenie.
 |
| Kwas perfluoromasłowy |
Wpływ takich związków na zdrowie jest słabo poznany ale prawdopodobnie są toksyczne dla tarczycy.
Problem z ich uwalnianiem polega na tym, że są bardzo trwałe. Wiązanie węgiel - fluor ma dużą trwałość i nie jest rozbijane przez mikroorganizmy, trudno też je rozerwać przez czynniki fizyczne. Rozpad długich cząsteczek zwykle zaczyna się od wymiany podstawnika przy węglu na inny, wchodzący w różnorodne reakcje. Jeśli cała duża cząsteczka organiczna jest "pokryta" podstawnikami fluorowymi, czynniki środowiskowe nie mają jak jej naruszyć. W związku z tym substancje takie trwają w środowisku niezmienione przez lata. Z drugiej strony ich usunięcie ze ścieków i odpadów przemysłowych jest trudne bo wymaga agresywnych warunków, które w dużej skali są drogie i niebezpieczne. Dla odpadów suchych i stałych główną metodą unieszkodliwiania jest spalanie, co ze względu na powstający fluorowodór wymaga specjalnej aparatury. Dla odpadów wodnych zawierających niewielką ilość PFAS jest mniej dostępnych technik. Testowane było w ostatnich latach rozwiązanie wykorzystujące wodę w stanie nadkrytycznym do utleniającej degradacji, przy wysokim ciśnieniu i temperaturze 400 stopni. Zamiast tego gromadzenie odpadów płynnych i stałych z tymi związkami w beczkach i cysternach to nie jest rozwiązanie. Dlatego najnowsze odkrycie, że można je zdegradować do nietoksycznych produktów w dość łagodnych warunkach daje szansę na zmniejszenie uwalniania do środowiska.
Grupa badaczy z USA i Chin skupiła się na związkach perfluorowanych z grupą karboksylową na jednym z końców. Jest to w zasadzie jedyny punkt, w którym cząsteczka może z czymś reagować. Postanowiono wykorzystać znaną już wcześniej reakcję dekarboksylacji w warunkach zasadowych pod wpływem wodorotlenku sodu. Oczekiwano, że dojdzie do odszczepienia grupy -COOH, pozostanie perfluorowany ogon podstawiony grupą alkoholową, z możliwością utleniania i odczepiania węgla po węglu.
Efekty podczas pierwszych prób przeszły jednak oczekiwania. Zaszła szybka degradacja do małocząsteczkowych produktów. Z długiego łańcucha zaczęły nagle odpadać kolejne fluoru a pozbawiony ich ochrony związek utleniał się i rozpadał tworząc produkty z jednym, dwoma i trzeba atomami węgla. Bardzo ciekawe. Właściwie sprzeczne z teorią. Potrzeba było wielu analiz związków pośrednich i obliczeń mechanizmów prowadzących do postawienia i defluoryzacji aby zrozumieć co takiego dzieli cząsteczki od razu na kilka kawałków.
Wydedukowano a potem potwierdzono przez wykrycie związków pośrednich mechanizm który za to odpowiada. W pierwszym etapie następuje normalna dekarboksylacja jak to oczekiwano. Tuż po usunięciu grupy karboksylowej w jej miejscu na chwilę pozostaje ładunek ujemny. Powstaje karboanion organiczny. Chętnie łączy się on z dowolnym protonem jaki tylko znajdzie i w szybkiej reakcji tworzy związek z jednym wodorem zamiast fluoru. Jednak w takich warunkach jak prowadzone i takim rozpuszczalniku karboanion ma większą trwałość. Część cząsteczek zostaje w takiej formie a te które złapały jakiś proton zaraz go tracą z powodu jego kwasowości (równowaga reakcji jest silnie przesunięta). Skoro więc dużo związku pozostaje dłużej w takiej formie, jest czas aby zaszedł proces znany z chemii węglowodorów - ładunek ujemny wędruje po cząsteczce tworząc i zrywając wiązania. Następujące przegrupowanie tworzy wiązania podwójne węgiel - węgiel przez co konieczne jest odrzucenie fluoru. Powstający nienasycony związek nie jest już taki odporny na reakcję. Następuje przyłączenie grupy OH do wiązania podwójnego z powstaniem kolejnego karboanionu. A ten jest stabilizowany przez warunki, zachodzi przegrupowanie, odszczepienie fluoru, powstanie wiązania podwójnego, które reaguje z grupą OH... I tak dalej wiele razy aż łańcuch popęka. Ostatecznie głównymi produktami degradacji jest dwutlenek węgla, szczawiany sodu, jony fluorkowe i fluorooctan sodu. A z ich usunięciem już umiemy sobie radzić.
 |
| Proponowany mechanizm degradacji z suplementów do publikacji |
Etapem limitującym szybkość reakcji jest początkowa dekarboksylacja, zachodząca w temperaturze 120 stopni. Startując z pierwszego związku pośredniego z jednym atomem azotu, dalszą degradację da się przyprowadzić w temperaturze 40 stopni.
Wiązania w benzenie poprawione
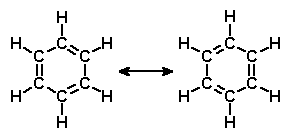
Benzen to jedna z najważniejszych cząsteczek w historii chemii organicznej. Na jego przykładzie rozwiązano kilka ważnych problemów związanych z budową cząsteczek, jego pierścień jest też składową wielu cząsteczek naturalnych lub syntetycznych. Dlatego ważne jest aby znać jego właściwości jak najdokładniej. Ostatnie badania naukowców z Korei Południowej dołożyły jeszcze jedną cegiełkę. Normalny benzen zawiera sześć atomów węgla połączonych z sześcioma atomami wodoru. Wodór jednakowoż występuje naturalnie w odmianach izotopowych, jako prot i deuter, chemicznie identycznych ale różniących się masą i odrobinę właściwościami fizycznymi. Dotychczasowe badania sugerowały, że w obu wersjach długości wiązań C-H i C-D są takie same i tak to przedstawiała literatura. Z drugiej jednak strony, spodziewać się można było jakiegoś jednak efektu izotopowego. W końcu różnica masy jądra między deuterem a protem jest aż dwukrotna, więc równowagowe, średnie położenie takiej masy na końcu oscylującego wiązania powinno być nieco inne.
W nowszym badaniu użyto specjalnej, odpowiednio dostrojonej odmiany analizy widm Ramana, pozwalającej badać właściwości wiązań dzięki obserwacji rozpraszania światła na rotujących cząsteczkach. Dokładność określenia długości wiązań została dzięki temu znacznie poprawiona i udało się znaleźć różnicę między wiązaniami - to z deuterem jest o 11,5 mÅ krótsze niż to z wodorem. Efekt jest bardziej zgodny z obliczeniami kwantowymi niż dotychczas.

Potencjalnie bardziej precyzyjne dane o tym ile wynosi długość tego wiązania w cząsteczkach aromatycznych poprawi jakość symulacji dynamiki molekularnej lub jakość udokładnienia struktury krystalograficznej związków deuterowanych.
* Mass-Correlated High-Resolution Spectra and the Structure of Benzene, I Heo et al, J. Phys. Chem. Lett., 2022, 13, 8278 (DOI: 10.1021/acs.jpclett.2c02035)



































{kind=link}