
Jak działa mikrofalówka?
Mikrofale są falami promieniowania elektromagnetycznego, podobnie jak fale radiowe czy światło widzialne. Oznacza to, że w punkcie przestrzeni przez który przechodzą, pojawia się pole elektryczne i magnetyczne, których wektor natężenia (kierunek w którym następuje zmiana) zmienia zwrot co pewien okres, zależny od częstotliwości.
Dość arbitralnie uznaje się za mikrofale, falę o długości (odległość w przestrzeni między dwoma punktami o takim samym wektorze pola) od 1 mm do 30 cm. Niosą więcej energii niż dłuższe fale radiowe, ale mniej niż krótsza podczerwień.
Ogrzewanie przez nie ciał następuje wskutek oddziaływania z dipolami. Jeśli cząsteczka związku chemicznego ma nierównomiernie i niesymetrycznie rozłożony ładunek elektryczny, to nabiera momentu dipolowego, to jest zachowuje się jak układ leżących blisko ładunku dodatniego i ujemnego. Następuje to wtedy, gdy tworzą ją atomy różniące się elektroujemnością, to jest zdolnością do przyciągania elektronów, i nie są ułożone tak, że posiadają środek symetrii. W przeciwnym przypadku części cząsteczki powtarzające się po obu stronach tegoż środka ściągałyby ładunek w przeciwne strony i nie było by żadnego wyróżnionego kierunku.

Dipole, które zachowują się jak układ przeciwnych ładunków, reagują na pola elektryczne lub magnetyczne. W stałym, dostatecznie silnym polu, będą się obracać równolegle do wektora jego siły, tak aby do źródła mającego ładunek dodatni lub ujemny obrócony był fragment cząsteczki o przeciwnym ładunku.
Podobnie rzecz się ma gdy na dipol oddziałuje fala elektromagnetyczna - cząsteczka wpada w zmienne pole, którego wektor co chwila zmienia się, wskazując raz to w jedną raz to w przeciwną stronę. W przypadku fal krótkich, jak dla światła widzialnego, gdzie długość fali jest liczona w nanometrach, czas przez jaki cząsteczka omiatana falą znajduje się w polu o jednym kierunku jest tak krótki, że nie nadąża zareagować. W przypadku długich, jak fale radiowe, wprawdzie pozostaje ona w polu danego kierunku długo, ale sama fala niesie ze sobą niską energię. Pośrodku znalazły się mikrofale - na tyle długie, aby cząsteczka zdążała zareagować, i zarazem niosące dostatecznie dużo energii aby po jej otrzymaniu cząsteczka pokonała swoją bezwładność.

W skrócie - cząsteczki będące dipolami w polu elektrycznym będącym składową mikrofal, zaczynają drgać. A drgania całych cząsteczek to właśnie to, co fizycy nazywają temperaturą. Energia kinetyczna rozedrganych mikrofalami cząsteczek dipola rozchodzi się przez oddziaływanie z innymi cząsteczkami. Mikrofalówki domowe posiadają źródła mikrofal o częstotliwości 2,45 GHz, co odpowiada długości fali 12,2 cm. Fala tej długości działa przede wszystkim na cząsteczki wody, posiadające wyraźny moment dipolowy. To od wody rozgrzewa się całe danie. Fale wnikają wgłąb porcji na 2-3 cm, co w zasadzie oznacza, że przenikają ją całą od momentu włączenia.
Ten typ ogrzewania różni się od konwencjonalnego przede wszystkim szybkością - w przypadku gotowanej kiełbaski rozchodzenie się ciepła następuje od zewnątrz do środka. Najpierw zużywamy ciepło na zagotowanie wody. Potem woda przekazuje energię zewnętrznym warstwom kiełbaski, te poprzez oddziaływania cząsteczek przekazują energię warstwom głębszym i tak dalej aż całość ogrzeje się na tyle, aby dało się to zjeść. Bywa że większe kawały mięsa zostają niedogotowane w środku. To rozchodzenie się ciepła nie jest zbyt szybkie, praktycznie żadnej roli nie pełni tu konwekcja wody wewnątrz jedzenia, mięso czy tkanki roślinne zwykle nie są zbyt dobrymi przewodnikami ciepła. Trzeba więc utrzymywać w stanie wrzenia wodę z naszą kiełbaską odpowiednio długo.
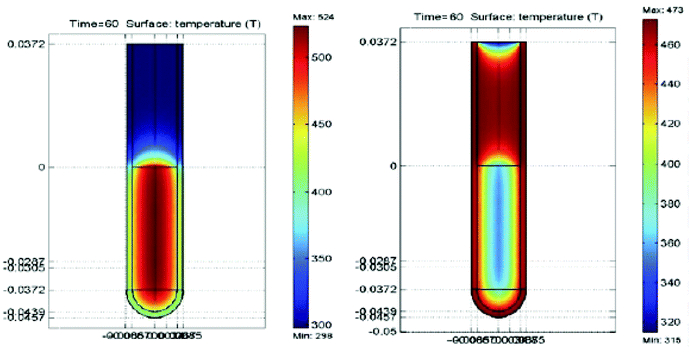 |
| Ogrzewanie cieczy w próbówce, po lewej przy pomocy mikrofal a po prawej przy pomocy łaźni olejowej. Po 60 sekundach ogrzewania w mikrofali zawartość próbówki jest ciepła, bardziej wewnątrz, a w łaźni ogrzało się szkło próbówki i przylegająca warstwa cieczy |
Mikrofale przenikają porcję jedzenia od razu od momentu włączenia, a ciepło jest wytwarzane w środku, przez co uzyskanie tej samej temperatury następuje krócej. Jedzenie jest wręcz cieplejsze w środku niż na zewnątrz, ze względu na wypromieniowanie ciepła przez powierzchnię, mającą kontakt z wcale nie gorącym powietrzem. Ma to pewien paradoksalny skutek, możliwy do zaobserwowania w doświadczeniu z kostkami sera - na talerz mikrofalówki kładziemy kostki żółtego sera, jedną dużą drugą mniejszą. Po włączeniu na średnią moc możemy zauważyć, że większa kostka stopi się szybciej.
Fale używane w kuchence mikrofalowej mają długość 12,2 cm. Są zatrzymywane przez metalową siatkę widoczną w szkle drzwiczek, ale obecną też w ściankach, której oczka są dużo mniejsze.
Utrata witamin
Teksty temat tego jak bardzo złe jest ogrzewanie jedzenia w mikrofalówce najczęściej powtarzają twierdzenie, że mikrofala "wyjaławia żywność" lub "niszczy wszystkie składniki odżywcze". W tym wyjaławianiu coś jest na rzeczy, bo podgrzanie jedzenia zwykle zabija bakterie, a w tym przypadku równomierne rozgrzanie od środka rozwiązuje problem niedosmażenia mięsiwa. Jak jednak wygląda sprawa składników odżywczych? W końcu takie na przykład witaminy są często wrażliwe na obróbkę cieplną.
Podczas ogrzewania część witamin bądź rozkłada się, bądź utlenia. Na to jak dużo substancji przereaguje, oprócz temperatury wpływ ma też czas trwania procesu. Jeśli w mikrofali osiągnięcie tego samego podgrzania następuje szybciej, można spodziewać się mniejszych strat witamin. I to pokazały badania. Znalazłem obszerny polskojęzyczny przegląd prac na ten temat.
Dla witaminy C stwierdzono, że ogrzewanie w mikrofali brokułów wywołuje mniejsze straty niż podczas zwykłego gotowania, porównywalnie małe dawało gotowanie pod ciśnieniem. Gotowanie tak ziemniaków, kalafiora, brukselki i fasolki szparagowej dawało straty znacząco mniejsze niż dla gotowania zwykłego. [1]
Dla witaminy B1 obserwowane straty przy gotowaniu mikrofalowym były bądź porównywalne (dla ciecierzycy) lub nieco mniejsze (dla boćwiny i zielonej fasolki) niż w zwykłym gotowaniu. Dla witaminy B2 straty w mikrofali były porównywalne lub mniejsze, z wyjątkiem zielonego groszku gdzie były większe. Dla witaminy PP straty w gotowanej ciecierzycy były mniejsze. [2]
Polifenole
Do najczęściej powtarzanych argumentów należy niszczenie polifenoli i flawonoidów podczas ogrzewania w mikrofali. Niekiedy artykuły powołują się na konkretne badanie, w którym stwierdzono, że w gotowanych tak brokułach zawartość tych związków spada aż o 97%. Faktycznie, takie dane pojawiają się w cytowanym badaniu. Artykuły jednak nie są chętne wejść w szczegóły - brokuły były w tym badaniu gotowane w wodzie przy pomocy mikrofal, większość strat polifenoli wynikała nie z rozkładu tylko z rozpuszczenia się ich w wodzie, którą wylewano. W czasie tego samego badania stwierdzono też stratę 66% polifenoli podczas gotowania tradycyjnego.[3]
Sami autorzy przyznają zresztą, że te wyniki znacząco odbiegają od uzyskanych w innych badaniach, przykładowo japońscy badacze badający gotowanie cebuli stwierdzili, że między gotowaniem tradycyjnym a mikrofalowym nie ma różnicy dla strat polifenoli [4] a indyjski zespól badający w ten sposób 14 warzyw stwierdził, że straty podczas gotowania na parze i w mikrofali były mniejsze niż podczas gotowania zwykłego.[5]
Tak, że no owszem, bardzo dużą stratę zawartości polifenoli stwierdzono, ale tylko w jednym badaniu. Ponieważ główną przyczyną strat było rozpuszczanie polifenoli w wodzie którą potem odlewano, można się domyśleć, że w przypadku potraw których część stanowią warzywa, i z których nie odlewa się wody, straty będą bardzo małe.

Zmiana kształtu białek
Kolejnym częstym argumentem, jest zmienianie kształtu białek i powodowanie, że stają się nienaturalne. A jeśli coś jest nienaturalne to organizm tego nie poznaje i nie trawi.
Jest to kolejna wieść z kategorii "coś dzwoni ale nie wiadomo w którym uniwersytecie". Owszem, ogrzewanie żywności w mikrofalówce zmienia struktury białek, i to na takie których w naturze nie ma. Tylko że nie jest to nic nadzwyczajnego, zachodzi też podczas smażenia i gotowania. Proces ten nazywa się denaturacją. Przykładowo podczas smażenia jajecznicy rozpuszczalne w wodzie albuminy, będące białkami o cząsteczkach w kształcie kłębków sznurka, rozwijają się i prostują, po czym tworzą siatkę wielu przeplecionych łańcuchów. Taka struktura nie występuje w naturze. Jednak organizm nie ma z nią żadnego problemu. Nasz żołądek nie trudni się rozpoznawaniem struktury białka i testowaniem naturalności konformacji, tylko wytwarza enzymy które tą strukturę naturalną czy nie, niszczą i trawią.
Izomeryzacja aminokwasów
W paru miejscach widziałem, że dla udowodnienia szkodliwego wpływu powoływano się na badania na temat izomeryzacji aminokwasów.[6] Jak to już było tłumaczone we wpisie o witaminie C, w pewnych przypadkach asymetria cząsteczki powoduje, że związki chemiczne mogą posiadać dwie formy, podobne jak lustrzane odbicia ale nie nakładające się.

No i otóż, jak wykazano podczas ogrzewania mleka w mikrofali, część wolnych aminokwasów ulega izomeryzacji w nienaturalną formę D. Co to powoduje? Wedle artykułów szkodzi i ma uzasadniać szkodliwość mikrofalówek.
Czy D-aminokwasy, które normalnie nie wchodzą w skład białek żywych organizmów, nie występują w przyrodzie i pojawiają się dopiero po nienaturalnym ogrzaniu mikrofalami? Nie.
Nie są zbyt częste, ale pojawiają się w organizmach i żywności wskutek spontanicznej izomeryzacji. Reakcja taka, jak wiele innych, ma pewną określoną szybkość, która zwiększa się po ogrzaniu. I to zupełnie niezależnie od sposobu ogrzewania.
W pewnym badaniu ogrzewano mleko bądź w mikrofalówce lub konwencjonalnie i zbadano zawartość izomerów D kwasu asparaginowego i glutaminowego. Przed ogrzaniem mleko zawierało około 0,4-0,45% tych izomerów, po ogrzaniu zarówno mikrofalowym jak i zwykłym ich zawartość zwiększyła się o 0,25%. Między próbkami ogrzewanymi na różne sposoby przez ten sam czas nie było różnic w zawartości D-izomerów.[7]
W zasadzie z uwagi na to, że mikrofalami można ogrzać mleko szybciej, można by się spodziewać mniejszych poziomów tych izomerów.
Wspomniane izomery D nie są tak znów nienaturalne, skoro dokładne badania wykazują ich niewielkie ilości w różnych produktach naturalnych. W obszernym przeglądzie badań stwierdzono na przykład, że izomery aminokwasów białkowych występują zarówno w mleku surowym jak i pasteryzowanym. Większe ilości pojawiają się w produktach fermentowanych, a więc kefirze, jogurcie i serach, zwłaszcza w serze długo dojrzewającym, wskutek metabolizmu bakterii. Śladowe ilości pojawiają się w mięsie kurczaka i bekonie, i zwiększają po smażeniu, a nawet w chlebie gdzie podpieczenie tosta zwiększa poziom D-aminokwasów prawie dwa razy.[8]
Wykrywa się je także w ludzkim organizmie i podejrzewa, że mogą pełnić pewne funkcje biologiczne, być na przykład neuroprzekaźnikami, trudno więc straszyć ich obecnością.
Enzymy
W paru artykułach wspomniano o niszczeniu przez mikrofale enzymów z jedzenia. To prawda, ale przyczyną jest samo podgrzanie. Enzymy są białkami a wysoka temperatura zmienia ich strukturę i je unieczynnia. Nie ma znaczenia w jaki sposób odbywa się ogrzanie.
Nitrozoaminy
Nitrozoaminy to rakotwórcze produkty reakcji zachodzących w wysokiej temperaturze między aminami i azotynami. Pojawiają się w przetworzonym mięsie podczas pieczenia i smażenia. Artykuły o mikrofalówkach często twierdzą, że podczas ogrzewania w nich tworzy się konkretnie d-nitrozodietanoloamina (NDMA), trudno powiedzieć czemu akurat ta. Są badania na temat powstawania nitrozoamin w ogóle. Na przykład porównując boczek pieczony w mikrofali ze smażonym na patelni, stwierdzono powstawanie nitrozoamin w obu, w tym smażonym w większej ilości.[9]
W badaniu zawartości NDMA w koreańskich owocach morza ogrzewanych na sześć różnych sposobów, stwierdzono że najwyższe poziomy tej substancji powstawały podczas ogrzewania na grillu na węgiel drzewny, a najmniejsze przy ogrzaniu w mikrofalówce i gotowaniu w ciśnieniowej kuchence parowej.[10]

Inne dziwaczne argumenty
Artykuły o mikrofalówkach często są doprawione dziwacznymi wieściami, które w zasadzie nie stanowią żadnych argumentów, ale mają robić atmosferę. Do najczęściej powtarzanych należy wiadomość że ZSRR zakazało domowych mikrofalówek ze względu na dbanie o zdrowie obywateli, tylko nie wiadomo kiedy - daty wahają się od lat 50 do lat 80. Czasem pojawia się twierdzenie, że wynaleźli ją Naziści.
Inną często przytaczaną opowieścią jest śmierć pacjenta któremu przetoczono krew, wcześniej podgrzaną w mikrofalówce, oczywiście wskutek szkodliwego wpływu mikrofal na krew. Taka historia faktycznie miała miejsce w 1989 roku w stanie Oklahoma w USA. Ale przyczyna śmierci była inna - pielęgniarka miała ogrzać krew przechowywaną w lodówce do temperatury ciała pacjenta. Zamiast użyć standardowej łaźni z ciepłą wodą, użyła mikrofali, określając potrzebny czas na wyczucie. W efekcie krew zamiast ogrzać się do tych około 36 stopni, przegrzała się. Wprawdzie ochłodzono ją ale z powodu za wysokiej temperatury doszło do częściowej hemolizy, czyli pęknięcia czerwonych krwinek. Po wprowadzeniu jej do krążenia pojawiły się skrzepy, będąc przyczyną śmierci pacjentki. W 1995 roku rodzina zmarłej dostała odszkodowanie.[11]
Do tego dochodzą takie dziwne twierdzenia jak gromadzenie się mikrofal w jedzeniu, wodzie a nawet meblach kuchennych (!).
Prawdziwy problem
Prawdziwy problem w żywności z mikrofalówki jest taki, że niezbyt dobrą jakość ma żywność do niej wkładania. Gotowe zestawy do podgrzania to często już na wstępie mocno przetworzona żywność z dużą ilością tłuszczy, małą błonnika i witamin. Nie powinny stanowić podstawowej diety w ciągu dnia.
--------
[1] http://www.ptfarm.pl/pub/File/Bromatologia/2013/3/BR%203-2013%20s.%20241-249.pdf
[2] https://www.ptfarm.pl/pub/File/Bromatologia/2013/3/BR%203-2013%20s.%20250-257.pdf
[3] http://www.ingentaconnect.com/content/jws/jsfa/2003/00000083/00000014/art00018;jsessionid=1smkxaf0ymund.victoria
[4] https://www.ncbi.nlm.nih.gov/pubmed/11349895
[5] http://www.ijfans.com/vol2issue3/9.pdf
[6] https://www.ncbi.nlm.nih.gov/pubmed/1968186
[7] https://www.ncbi.nlm.nih.gov/pubmed/7911811
[8] http://www.acta.sapientia.ro/acta-alim/C2-1/alim2-1.pdf
[9] https://www.ncbi.nlm.nih.gov/pubmed/2307266
[10] http://www.tandfonline.com/doi/abs/10.1080/0265203021000014770
[11] http://wyomcases.courts.state.wy.us/applications/oscn/DeliverDocument.asp?citeID=4387












{kind=link}