Więc...
Dotarłem już w nauce akademickiej do tego momentu, gdy zamiast poprzestawać wyłącznie na powtarzaniu już opisanych i przygotowanych reakcji, muszę zacząć podjąć własną pracę naukową - oczywiście pod bacznym okiem promotora, dr Ewy Wolińskiej.
Dokładnie określonego tematu pracy jeszcze nie mam, ale zasadniczo opierać się będzie ona na syntezie zawierających 1,2,4-triazynę ligandów, do katalizatorów mających posłużyć do syntezy asymetrycznej. Zanim jednak omówię coś z przeprowadzonych syntez, muszę oczywiście objaśnić co też są to te triazyny, ligandy i dlaczego synteza miałaby być asymetryczna; ponieważ jednak objaśnienia te bardzo mi się wydłużyły, uznałem że ten wstęp teoretyczny podam w jednym wpisie, zaś trzy kolejne etapy właściwej syntezy omówię w kolejnych. Teraz więc będzie o tym, czy mogą istnieć "lewe" cząsteczki i jak to się ma do naszego zdrowia:
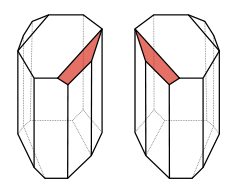
Jedną z właściwości cząsteczek organicznych, jest posiadanie określonej symetrii. Popatrzcie na swoje ręce; najlepiej wyciągnijcie je przed siebie i połóżcie jedną na drugą, bez obracania ku sobie. Nie nakładają się. Kciuki sterczą w przeciwne strony. Możemy jednak obrócić jedną i złożyć z drugą jak do modlitwy, wtedy ich obrysy będą się nakładały, ale nadal nie będą identyczne, bo ich grzbiety będą skierowane w przeciwne strony. Jak byśmy ich nie obracali, jedna nie stanie się taka jak druga.
To zupełnie oczywiste - jedna jest dłonią lewą a druga prawą. Są do siebie podobne jak lustrzane odbicia. Gdyby ktoś miał idealnie symetryczne dłonie, to lustrzane odbicie jednej wyglądałoby dokładnie tak jak druga.
O bryłach mających tą właściwość, że podobnie jak dłonie, posiadają formę "lewą" i "prawą", które nie dają się na siebie nałożyć przez obrót w przestrzeni, i są do siebie podobne jak lustrzane odbicia, mówimy że są chiralne (od greckiego
chira - ręka). Jest wiele takich figur. Istnieją lewe i prawe muszle ślimaków, kwiaty, a z przedmiotów codziennego użytku, nożyczki dla prawo i leworęcznych:
Nie inaczej jest ze związkami chemicznymi. Już na początku XIX wieku skrupulatny badacz Ludwik Pasteur, najbardziej znany z badań nad fermentacją, przyglądając się kryształom soli syntetycznego kwasu winowego zauważył, iż są one asymetryczne, oraż że tworzą dwie odmiany, podobne jak lustrzane odbicia. Gdy zaś oddzielił jedną odmianę od drugiej, po prostu sortując kryształki pincetą, stwierdził że kwas winowy "prawy" zawsze krystalizuje w takiej formie. Uznał więc, że widocznie musi istnieć prawa i lewa odmiana kwasu winowego, które różnią się kształtem cząsteczki. Teoria atomowa była wówczas w powijakach, a co dopiero teoria struktury cząsteczek, toteż przez długi czas ta luźna hipoteza nie znajdowała zainteresowania. Do czasu gdy odkryto wreszcie jak rozłożone są w przestrzeni wiązania z węglem w związkach organicznych.

Węgiel w tych związkach tworzy cztery wiązania z innymi atomami - mogą to być wiązania potrójne, podwójne lub pojedyncze. Ponieważ każde wiązanie stanowi parę elektronową, a każda taka para odpycha się od innej, starają się one rozłożyć w przestrzeni w największym możliwym oddaleniu, co w przypadku czterech wiązań pojedynczych realizuje się w formie rozłożenia tetraedrycznego - to jest atom znajduje się jakby w środku foremnego czworościanu, a wiązania biegną do naroży. Jeśli teraz zdarzy się, że przy każdym z wiązań podczepiona będzie inna grupa, to cała cząsteczka stanie się chiralna, i możliwe staną się dla niej dwie konfiguracje - lewa i prawa, podobne jak lustrzane odbicia, jak to widać na tej pięknej grafice:

Związki chemiczne mające taką właśnie lustrzaną właściwość, to enancjomery, zaś atom węgla (czasem może to być fosfor lub azot, ale rzadziej) wokół którego pojawia się ta szczególna asymetria, nazywany jest atomem asymetrycznym, centrum chiralnym lub też jak zaleca się ostatnio centrum stereogenicznym. Konfigurację podstawników wokół takiego atomu określa się na różne sposoby - najczęściej używana metoda, polega na przypisaniu podstawnikom "ważności", tak że na przykład grupa metylowa jest ważniejsza od podstawnika wodorowego, etylowa od metylowej, a chlorkowa od etylowej. Jeśli teraz tak obrócić nasz asymetryczny atom, aby podstawnik o najmniejszej ważności znalazł się z tyłu, to konfigurację określa kierunek w którym poustawiane są pozostałe - gdy od najważniejszego do najmniej ważnego ruch jest zgodny z kierunkiem wskazówek zegara, to konfiguracja jest określana literą R, gdy jest odwrotnie, konfigurację określamy S. Są też inne typy konfiguracji, na przykład dla cukrów i aminokwasów zwykle używa się oznaczeń D i L.

Gdy związek ma więcej jak jedno takie miejsce, sytuacja się komplikuje, bo wówczas możliwa jest większa liczba kombinacji - każde centrum może mieć dwie konfiguracje. Kwas winowy ma dwa takie miejsca, stąd możliwe są dla niego trzy odmiany: gdy oba centra mają konfigurację R, gdy oba mają S i gdy jedno ma R a drugie S. Glukoza ma cztery takie miejsca i dla niej możliwych jest 16 odmian, cholesterol ma 8 takich miejsc i teoretycznie mógłby mieć ponad 200 odmian, choć niektóre struktury za bardzo deformowałyby cząsteczkę. Takie odmiany wielocentrowe, nazywamy stereoizomerami, i nie są one już swymi lustrzanymi odbiciami.
Natomiast mieszaniny równych ilości R i S izomerów, nazywamy racematami.
I co z tego?
Izomery różniące się konfiguracją, mają takie same właściwości chemiczne, jednak dość istotne różnice zachodzą w ich oddziaływaniu biologicznym, oto bowiem my sami jesteśmy chiralni.
Podstawowymi związkami strukturalnymi organizmów żywych są białka, te zaś zbudowane są z aminokwasów - związków, zawierających grupę aminową i karboksylową, połączonych do tego samego węgla. Jeśli dwa pozostałe podstawniki są różne, to cząsteczka staje się chiralna i tak właśnie jest w przypadku wszystkich biogennych aminokwasów, z wyjątkiem glicyny. Z tych chiralnych cząsteczek zbudowane są białka, a z białek elementy strukturalne, i jak się okazuje, bardzo często konfiguracja substancji wpływa na reakcje jakim ulega w naszym organizmie. Jednym z takich znanych przypadków, jest
limonen - izomer D ma zapach pomarańczy i występuje w skórce tego owocu, izomer L ma zapach terpentyny i występuje w roślinach szpilkowych. Będący jego pochodną
karwon ma jeszcze wyraźniejsze różnice zapachu - izomer S pachnie anyżkiem, a izomer R miętą. Inny terpenoid,
mentol, ma trzy centra i 8 odmian; odmiana występująca w mięcie polnej i mająca najsilniejsze działanie i zapach, ma konfigurację 1R,2S,5R, pozostałe występują rzadko lub zostały otrzymane sztucznie
W podobny sposób różnią się smaki izomerów - lustrzane wersje substancji słodkich mogą mieć smak kwaśny lub gorzki, choć nie zawsze tak jest. Lustrzana wersja glukozy jest tak samo słodka jak oma, ma jednak jedną ciekawą właściwość - nie pasuje do pierwszego enzymu, rozpoczynającego metabolizm. Powoduje to, że nie jest przetwarzana na energię i zostaje w niezmienionej formie wydalona - byłaby zatem idealnym słodzikiem, słodkim ale nie kalorycznym. Niestety jej produkcja jest nieopłacalna.
Dla nas jednak najistotniejszą kwestią nie jest smak czy zapach, lecz działanie na organizm. Nie zawsze, ale jednak bardzo często to, czy dana substancja będzie dla organizmu obojętna, szkodliwa czy lecznicza, zależy od konfiguracji jej centrów stereogenicznych, jeśli takie posiada. Przykładowo lek przeciwbólowy
Ibuprofen jest zwykle syntezowany w formie racematu, jednak właściwości lecznicze ma tylko S izomer, co znaczyłoby, że połowa wyprodukowanego związku jest zupełnie niepotrzebna. Okazało się jednak że obie odmiany mogą zamieniać się w siebie w organizmie. Podobnie jest z
Naproksenem - tylko jeden izomer ma właściwości przeciwbólowe, a oba są toksyczne dla wątroby.
Niekiedy działanie odmian może być skrajnie różne, zależnie od konfiguracji.
D-propoksyfen jest środkiem przeciwbólowym; L-odmiana ma silniejsze działanie przeciwkaszlowe, ale w wysokich dawkach. Obie odmiany wycofano z powodu częstych sercowych skutków ubocznych. Naturalny kwas L-askorbinowy jest witaminą C, i bierze udział w pewnych przemianach enzymatycznych; izomer D jest nieaktywny i nie może być nazywany witaminą - choć też jest przeciwutleniaczem. Dlatego też witaminę syntetyczną produkuje się tak, aby otrzymać tylko L-izomer, w czym biorą udział pewne szczepy bakteryjne.
Amfetamina i
metamfetamina też mają dwie odmiany - odmiana D pobudza zarówno obwodowy jak i centralny układ nerwowy, i ma działanie narkotyczne; izomer L pobudza tylko OUN i nie wywołuje odurzenia, dlatego też ten izomer bywa stosowany w inhalatorach donosowych, wywołując skurcz naczyń krwionośnych. Inny środek narkotyczny,
nikotyna, w naturze występuje w odmianie S(-). Enancjomer R ma podobne działanie lecznice, ale jest znacznie mniej toksyczny - źródła podają że od 20 do 40 razy.
Skrajnym przypadkiem jest niechlubny
Talidomid, którego jeden enancjomer zapobiegał mdłościom, bólom głowy i miał działanie uspokajające, a drugi miał działanie teratogenne, uszkadzające płód. Produkowany preparat był racemiczną mieszanką obu izomerów, i zalecany kobietom w ciąży, co doprowadziło do narodzin tysięcy kalekich dzieci, co
kiedyś już opisałem. Obecnie bywa używany w chemioterapii do hamowania rozrostu guza.
Skoro między właściwościami izomerów istnieją na tyle istotne różnice, to chyba najlepiej byłoby wziąć tylko jeden z nich i stosować czysty związek? Jak najbardziej, tyle że nie jest to taka łatwa sprawa gdy mamy je zmieszane. Stereoizomery mają takie same właściwości fizyczne i chemiczne - jedynie czasem różnią się na przykład strukturą krystaliczną, lub szybkością reagowania i można próbować rozdzielać je w ten sposób. Zrobił to choćby Pasteur, sortując kryształki soli kwasu winowego, obie bowiem odmiany tego związku najchętniej tworzą kryształy zawierające tylko jedną z nich. Czynią to na tyle chętnie, że chemikowi udało się przeprowadzić bardzo zabawne doświadczenie - do kuwety z nasyconym racematem kwasu, włożył z jednej strony kryształek odmiany R a z drugiej odmiany S. Kryształy stopniowo rosły, przyjmując do sieci krystalicznej cząsteczki tylko jednej odmiany, takiej samej jak w krysztale zarodkowym, aż otrzymał dwa duże kryształy rozdzielonych izomerów.

Inny pomysł polega na zastosowaniu chiralnych reagentów, tworzących związki o wystarczająco różnych właściwościach. Przykładowo związek nasz w mieszaninie R i S jest lekko zasadową aminą, więc traktujemy go na przykład R,R kwasem winowym. Tworzą się nam dwie sole - RR-winian-R-aminy i RR-winian-S-aminy, które bardzo często różnią się rozpuszczalnością, bo chiralne fragmenty różnie ze sobą reagują. Chwytając kogoś prawą dłonią za prawą, możemy go złapać mocniej, niż prawą za lewą, i podobnie jest w tego typu solach.
Dla tych, gdzie oddziaływania są silniejsze, krystalizacja zachodzi chętniej, więc można je wydzielić przez wielokrotne przekrystalizowanie. Inny pomysł polega na tworzeniu estrów o różnej rozpuszczalności bądź temperaturze wrzenia. Bardziej wyrafinowane sposoby wykorzystują reakcje enzymatyczne - na przykład związek o naturze alkoholu przeprowadzamy w ester kwasu tłuszczowego i traktujemy którąś z esteraz - enzymów trawiennych przywykłych do rozkładania połączeń o jednej konfiguracji. Rozkład na przykład R-estru, daje nam selektywnie wyjściowy alkohol, czysty enancjomerycznie, możliwy do oddzielenia przez ekstrakcję. Jeszcze inna metoda polega na zastosowaniu chromatografii kolumnowej, z wypełnieniem zawierającym chiralne związki - na przykład krystaliczną celulozę - jest to jednak metoda bardzo droga.
A może łatwiej byłoby otrzymywać od razu jeden izomer, a nie mieszaninę dwóch? - w tym właśnie cały ambaras, aby nie powstawały oba na raz. Jeżeli poddajemy reakcjom związki już chiralne, i w trakcie reakcji centrum stereogeniczne nie jest naruszane, to otrzymamy selektywnie czysty izomer produktu, przykładowo redukując naturalne R-aminokwasy, otrzymamy R-aminoalkohole a z tych na przykład pierścieniową R-oksazolinę. Reakcje, gdy wychodząc z substratu o określonej konfiguracji, otrzymujemy produkt o określonej konfiguracji, nazywamy
stereoselektywnymi.
Nieco większy problem sprawiają nam reakcje, w których mamy stworzyć nowe centrum, wychodząc ze związku, który takiego nie posiada. Weźmy sobie taki prosty związek jak 1,3-dimetyloheksen, z jednym wiązaniem podwójnym. I poddajmy go reakcji przyłączenia chlorowodoru. Zgodnie z odpowiednimi prawami, wodór przyłączy się z tej strony wiązania, gdzie jest już drugi, a chlor przy grupie metylowej. I powstanie nam centrum stereogeniczne, mające w otoczeniu - przy jednym wiązaniu grupę metylową, przy drugim chlor, przy trzecim pierścień mający grupę metylową za 4 węgle a z czwartej strony ten sam pierścień, ale z grupą metylową za trzy węgle. Tylko jaka będzie konfiguracja? Mieszana.

Gdy atom chloru atakuje wiązanie podwójne, o płaskiej strukturze, może dotrzeć do cząsteczki z dwóch stron - od lewej i od prawej. Ponieważ cząsteczka jest płaska, szanse obu przebiegów są równe, w efekcie otrzymujemy równomolową mieszankę produktów, powstałych a ataku z lewej i z prawej, czyli R:S 1:1 - a zatem racemat.
Wszystkie metody syntezy, mającej zachwiać tą symetrią - a więc syntezy asymetryczne - opierają się na utrudnieniu dostępu z jednej strony, co może być osiągnięte na różne sposoby. Związek może być zaabsorbowany na powierzchni kryształu - jedna strona będzie zasłonięta i będzie się nam tworzył jeden produkt. Największe jednak zastosowanie mają specyficzne, chiralne katalizatory. Jak mogą działać?
Weźmy sobie cząsteczkę bardzo podobną do powyższej, ale z grupą hydroksylową, a więc 1-metyloheksen-3-ol. Grupa hydroksylowa przy trzecim węglu sama tworzy centrum stereogeniczne. Teraz przed dodaniem substraktu, używamy katalizatora - odpowiedniego kompleksu zawierającego jakiś metal, tak dobranego, że jon metalu może tworzyć wiązania koordynacyjne z elektronami Pi wiązania podwójnego, i wolnymi parami elektronowymi tlenu. Będzie zatem łączył się z cząsteczką od tej strony, z której jest grupa OH
zasłoni więc sobą jedną stronę, umożliwiając dostęp z drugiej strony. W tym przykładzie nowe centrum będzie miało konfigurację R. Jest to przykład wymyślony, ale pokazuje jak takie selektywne reakcje mogą zachodzić.
Prawdziwym majstersztykiem jest stosowana na skalę przemysłową synteza 1R,2S,5R-mentolu, a więc takiego samego związku jak naturalny. Związkiem wyjściowym jest terpenoid mircen, po katalitycznej izomeryzacji zamieniany na R-cytronellal a ten cyklizowany do ostatecznej cząsteczki z trzema centrami chiralnymi. Twórca tej metody
Ryoji Noyori w roku 2001 dostał nagrodę Nobla za prace nad asymetrycznym uwodornieniem.
Triazyny to związki organiczne, składające się z sześcioczłonowych pierścieni, w których znajdują się trzy atomy azotu. Możliwe są trzy ich ustawienia - w pozycjach 1,2,3, a więc wszystkie obok siebie; 1,2,4 - dwa obok siebie a jeden z odstępem; oraz 1,3,5 czyli symetrycznie rozdzielone. W moim przypadku zajmuję się 1,2,4-triazyną.
Pochodne triazyn dosyć chętnie tworzą kompleksy z jonami metali, i niektóre z nich mają zdolność do takiego katalizowania reakcji tworzących nowe centrum stereogeniczne, aby powstawał nadmiar jednego z izomerów, a co za tym idzie, zamiast racematu 1:1 otrzymujemy mieszaninę na przykład 6:4, 7:3 czy też najchętniej, ale rzadko 9:1 i wyższe.
A tym, czym będę się zajmował na pracowni, będzie tworzenie chiralnych ligandów do kompleksów mających wywoływać taką selektywność.