Niebieski
Gdy pies nasika chemikowi na ścianę...
Pittakall to prawdopodobnie pierwszy syntetyczny barwnik organiczny*, jaki pojawił się w handlu, choć nie zdobył zbyt dużej popularności i dość szybko zarzucono jego produkcję. Jego przypadkowym odkrywcą był niemiecki chemik Karl Reichenbach. Na początku XIX wieku zarządzając małą fabryką chemiczną zajął się badaniem produktów suchej destylacji drewna, węgla i szczątków organicznych. To on wydzielił ze smoły różne wartościowe frakcje, w tym kreozot, parafinę czy fenol, a także pierwszy olej opałowy nazwany eupinonem.
Kreozot, będący frakcją smoły z drewna drzew liściastych, miał dość charakterystyczną, silną woń oraz dobre właściwości konserwujące, dlatego chemik zaczął stosować go do impregnowania drewna. Do dziś zresztą jest częstym składnikiem impregnatów, na przykład do podkładów kolejowych.
Postanowił wypróbować go także w zastosowaniu dużo bardziej przyziemnym - przeszkadzało mu, że psy sikają mu zewnętrzną ścianę domu, więc posmarował ją kreozotem aby zapach je odstraszał. Psom najwyraźniej było wszystko jedno, bardzo lubiły tam stawać i podnosić nogę, lejąc bezczelnie po wysmarowanej ścianie. Cóż, widocznie nie było to dobry środek na psy.
Przyglądając się ich działalności zauważył jednak ciekawą rzecz - w miejscu gdzie stały plamy moczu, na ziemi pojawiło się wyraźne, niebieskie zabarwienie. A ponieważ był człowiekiem bardzo praktycznym, zaczął czynić próby powtórzenia reakcji. Szybko wykrył, że mocz nie jest w jej potrzebny, stanowił jedynie alkaliczny reagent. Prowadząc destylację surowej smoły stwierdził, że frakcja o temperaturze wrzenia wyższej niż kreozot, po wprowadzeniu do wody wapiennej lub roztworu wodorotlenku baru, po pewnym czasie zamienia się w ciemnogranatowy proszek.

Wprowadził go na rynek jako pigment nadający się do farbowania po rozpuszczeniu w alkaliach. Najwyraźniej jednak nie miał zbyt dobrych właściwości i po pewnym czasie przestał być używany, pojawiając się jedynie od czasu do czasu w historycznych spisach barwników. Dopiero pod koniec XIX wieku ustalono, że jest to związek będący produktem kondensacji pirogallolu, o strukturze podobnej do barwników trifenylometylenowych. W formie anionowej przybierał intensywny kolor. Pigment Reichenbacha był laką, to jest nierozpuszczalną solą barową lub wapniową.
Pittakall jest dziś w zasadzie historyczną ciekawostką. Mam wrażenie, że od ponad stu lat nikt go nie otrzymywał, bo poza wzmiankami w pracach o historii barwników nie znalazłem o nim żadnej dalszej informacji ani tym bardziej zdjęcia próbki. Ponoć miał dość ciemny odcień niebieskiego.[1]
Ftalocyjanina
Kolejny niebieski barwnik także został odkryty niezamierzenie, podczas otrzymywania czegoś innego, i to dwa razy.
W 1927 roku szwajcarscy chemicy Henri Diesbach i Edmond von de Weid zajmowali się znalezieniem lepszej niż już znane metody otrzymywania ftalonitrylu, to jest pochodnej benzenu z dwiema grupami -CN. Znana była już w tym czasie reakcja Sandmeyera, polegająca na podstawieniu soli diazoniowych, gdzie grupa -NN była łatwo zamieniana na inne. Przy jej pomocy otrzymywano ftalonitryl z o-aminobenzonitrylu (a ten z rozkładu termicznego amidu kwasu antranilowego).
Badacze postanowili spróbować nieco innej metody, której substrat był bardziej stabilny i łatwiejszy w otrzymaniu. Była to reakcja Rosenmunda-von Brauna (nie mylić z reakcją Rosenmunda samego, czyli redukcją kwasów do aldehydów) polegająca na podstawieniu halogenku przy pierścieniu aromatycznym, przez anion cyjankowy z cyjanku miedzi.[2]
Jako substratu użyli 1,2-dibromobenzenu. Pomysł był w istocie dosyć prosty, reakcja powinna przebiegać w taki oto sposób:
Produktem powinna być bezbarwna lub nieco żółtawa krystaliczna substancja. Jakież więc było zdziwienie chemików, gdy po reakcji znaleźli w kolbie osad intensywnie niebieski.
Produkt był bardzo trwały, nierozpuszczalny w wodzie i dość trudno w innych rozpuszczalnikach. Po wyznaczeniu przez analizę elementarną składu C26H18N6Cu uznali, że prawdopodobnie mają do czynienia ze związkiem kompleksowym ftalonitrylu i pirydyny, zawierającym jeden atom miedzi i po dwie cząsteczki tych związków [3]. Ale mylili się.
Drugimi odkrywcami byli chemicy w fabryce Scottish Dyes (dziś ICI), którzy w 1928 roku analizowali metodę przemysłowego otrzymywania ftalimidu. Bezwodnik ftalowy był w tej syntezie poddawany reakcji z amoniakiem w stężonym roztworze wodnym, zaś jako reaktorów używano emaliowanych żeliwnych kotłów. Mechaniczne mieszadło powodowało, że emalia z czasem się zdzierała, zaś pilnujący procesu technolodzy zauważyli, że partie produktu z tych najbardziej wytartych kotłów były zanieczyszczone drobnym, niebieskawym osadem. Było zresztą zauważalne, że na niebieskawy kolor zabarwił się odsłonięty metal.
Po zebraniu większej ilości zanieczyszczenia, pracownicy fabryki zdali sobie sprawę z tego, że potencjalnie mógłby to być niezły pigment, miał bowiem niską rozpuszczalność i bardzo dużą siłę barwiącą. Kolejną więc syntezę przeprowadzono dodając do masy wiórki żelazne, był to jednak proces mało wydajny. Należało odpowiedzieć na pytanie, co właściwie zachodzi w reaktorze i jaki związek otrzymano.
Po wpływem silnych alkaliów związek tracił metal, powstała wolna forma nadal była niebieska ale o dużo słabszym odcieniu. Można było połączyć ją z innymi metalami, zwłaszcza ze szczególnie chętnie wiązaną miedzią, tworząc kompleksy bardzo trwałe i intensywnie zabarwione. Analizy pokazały, że związek ma charakter aromatyczny i być może zawiera układ skumulowanych pierścieni. Dopiero w 1933 roku Patrick Linstead zaproponował dla związku budowę makrocykliczną, podobną do porfiryny, z czterema fragmentami benzopirolu połączonymi przez mostkowe azoty, co potwierdziły potem badania rentgenowskie.

Firma ICI zaczęła produkować pigment w 1934 roku po udoskonaleniu metod pod nazwą Monastral Blue, lub błękit ftalocyjaninowy. Był wielkim osiągnięciem. Niebieskich pigmentów było w tym czasie niewiele, w zasadzie istniały tylko nieorganiczne oparte o sole miedzi i ultramarynę, oraz indygo i jego pochodne. Błękit ftalocyjaninowy miał tą zaletę, że będąc barwnikiem organicznym posiadał wysoką odporność na blaknięcie, miał dużą siłę barwiącą, był na tyle słabo rozpuszczalny że nie migrował do innych warstw malarskich, oraz był dosyć odporny na czynniki fizyczne i chemiczne. Związek ten rozkłada się dopiero w temperaturze 600 stopni.
Do dziś jest jednym z najczęściej wykorzystywanych barwników, zwłaszcza do farb do metalu, ale też atramentów czy tuszu do długopisów. Jest na przykład składnikiem standardowego tuszu niebieskiego do drukarek, a ze względu na znikomą toksyczność także pigmentów do tatuażu.
Niezamierzony niebieski pigment
To odkrycie także miało pewien element przypadkowości.
Zespół profesora Subramaniana, na uniwersytecie stanowym w Oregonie, zajmował się badaniami tlenkowych materiałów ceramicznych z solami ziem rzadkich, które mogłyby potencjalnie znaleźć zastosowanie w elektronice. Niektóre mogłyby okazać się magnesami stałymi, inne nadprzewodnikami niskotemperaturowymi, ferroelektrykami czy superopornikami. Studenci profesora biorący udział w pracach testowali więc różne mieszanki tlenków i chlorków metali, które po wymieszaniu w młynie kulowym na bardzo drobny proszek wypalano w odpowiedniej temperaturze.
Doktorant Andrew E. Smith spróbował pewnego razu nietypowej mieszanki tlenku itru, indu i manganu VI. Dwutlenek manganu jest intensywnie czarny, dlatego po zmieleniu uzyskał ciemnoszary proszek. Następnie wsadził go na chwilkę do pieca aby się wygrzał. Temperatura szybko osiągnęła prawie 1100 stopni, a tlenki przereagowały ze sobą, tworząc nowe połączenie. Akurat to konkretne nie miało szczególnie ciekawych własności elektrycznych czy magnetycznych, lecz jedna właściwość rzucała się w oczy od razu po wyjęciu z pieca - otrzymany proszek okazał się niesamowicie niebieski.[4]
Gdy profesor zobaczył próbkę od razu przyszło mu do głowy, że to może znaleźć zastosowanie. Właściwie jeszcze przed publikacją na temat związku zaczęto starania nad komercjalizacją.
Jak się okazało podczas wyprażania powstaje związek zawierający warstwy tlenku manganu o nietypowej koordynacji w formie bipiramidy trygonalnej. W każdej takiej jednostce atom manganu otoczony jest przez pięć atomów tlenu, w tym trzy w płaszczyźnie warstwy i po jednym nad i pod nią. W takim położeniu oddziaływanie ligandów powoduje rozszczepienie poziomów energetycznych orbitali d manganu w taki sposób, że związek pochłania światło czerwone i zielone dając w efekcie niebieski kolor.

Wcześniej znany był pigment oparty o manganian baru, ale miał małą stabilność, nowy pigment nazwany YInMn nie tylko jest odporny na utlenienie czy redukcję, ale też zachowuje kolor w bardzo wysokich temperaturach i nie blaknie pod wpływem wilgoci. Szybko okazało się, że dobrze nadaje się zarówno do farb olejowych jak i wodnych, a także jako pigment do barwienia mas plastycznych. W tych zastosowaniach ważną własnością jest też jego nietoksyczność. Inne pigmenty nieorganiczne podobnej trwałości zwykle zawierają sole rakotwórczego kobaltu lub sole miedzi.
Podczas dalszych badań stwierdzono, że choć związek silnie pochłania światło czerwone, to zarazem silnie odbija podczerwień, osiągając jeden z najwyższych współczynników odbicia dla materiałów niemetalicznych (srebro odbija niemal 100% podczerwieni). Dzięki temu powierzchnie pomalowane farbą z tym pigmentem bardzo mało się nagrzewają, co miałoby znaczenie w przypadku na przykład dachów w cieplejszych krajach
Obecnie pigment zaczyna powoli wchodzić na rynek, niedawno producent kredek świecowych i pasteli Crayola ogłosił wprowadzenie kredki z YInMn, trwa konkurs na wymyślenie nazwy [5].
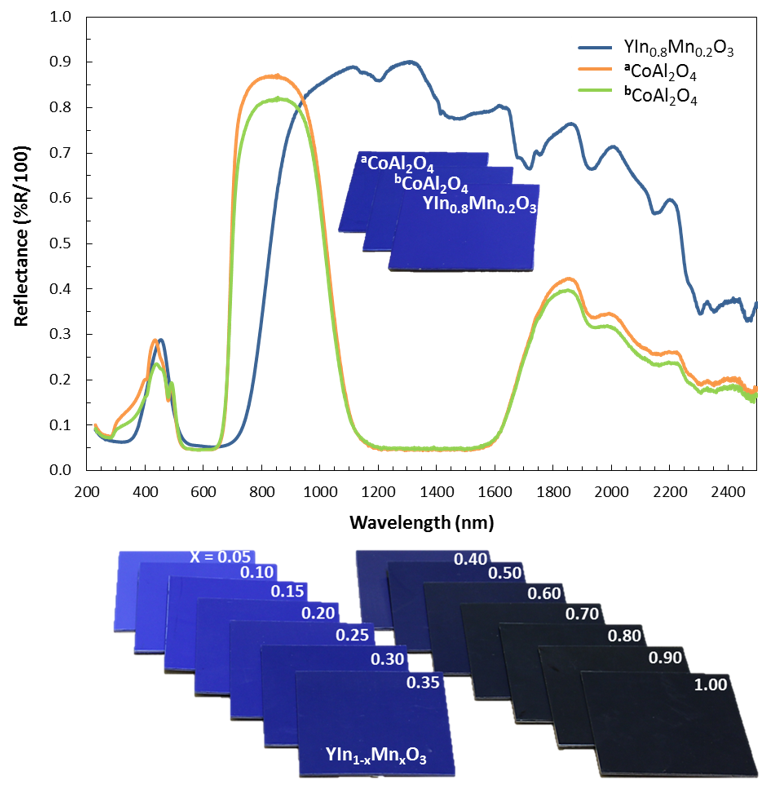
Jednak wbrew temu co piszą media, pigment nie posiada nowego, dopiero teraz odkrytego odcienia niebieskiego. W najbardziej optymalnym składzie YIn
0.8Mn
0.2O
3 , związek ma kolor błękitu kobaltowego lub nieco cieplejszy, mimo zupełnie różnego przebiegu krzywej absorpcji:
Odcień może płynnie zmieniać się w zależności od stosunku itru do manganu. Dodatek innych metali, na przykład tytanu czy cynku może natomiast zmienić kolor na zielony lub fioletowy.[6]
W zespole prof. Subramaniana trwają prace nad uzyskaniem pigmentu czerwonego o odcieniu i intensywności nie stosowanych dziś z powodu toksyczności pigmentów rtęciowych jak cynober.
Może jeszcze kiedyś napiszę coś o projektowaniu barwników aby pokazać, jak takie rzeczy otrzymuje się planowo, bez czekania na szczęśliwy przypadek.
-------
* Aczkolwiek wcześniej niż Pittakall bo w XVIII wieku stworzono kwas pikrynowy, przez pewien czas używany do farbowania wełny, zastosowanie jako barwnik znalazł jednak dużo później. Półsyntetyczny był natomiast otrzymany w podobnym czasie indygokarmin, znany jako błękit saksoński, pozyskiwany przez traktowanie indygo dymiącym kwasem siarkowym.
[1] George B. Kauffman,
Pittacal - The first synthetic dyestuff, Journal of Chemical Education (12) 1977 str. 753
[2]
Sandmeyer-von Braun reaction
[3] De Diesbach, Henri; von Der Weid, Edmond (1927).
"Quelques sels complexes des o-dinitriles avec le cuivre et la pyridine". Helvetica Chimica Acta. 10: 886.
[4] http://oregonstate.edu/ua/ncs/archives/2009/nov/accidental-discovery-produces-durable-new-blue-pigment-multiple-applications-0
[5] http://oregonstate.edu/ua/ncs/archives/2017/may/pigment-discovered-oregon-state-university-inspires-new-crayola-crayon-color
[6] http://chemistry.oregonstate.edu/content/story-yinmn-blue