Czyli dłuższa anegdota o odkryciu pewnego związku.
Wraz z rozwojem przemysłu w XIX wiecznej Europie, w tym maszyn parowych i pieców hutniczych, duże znaczenie jako paliwo zaczął odgrywać węgiel kamienny. Dla pewnych zastosowań korzystniejszym niż surowe paliwem był koks, otrzymany przez ogrzewanie węgla bez dostępu powietrza tak, że ulatywała zeń woda i lotnie związki. Koks, o wyższej wartości opałowej, zużywano głównie do wytopu stali; gazy palne zużywano do oświetlania ulic w latarniach i jako gaz do kuchenek; wykraplana woda pogazowa zawierająca amoniak była zużywana do produkcji nawozów sztucznych.
Jedynym produktem ubocznym jaki nie dawał się wprost zastosować była
smoła pogazowa, często po prostu wylewana albo po oddzieleniu najbardziej lotnych składników używana do impregnacji drewna. Szybko zainteresowali się nią chemicy świadomi, że jest mieszanką wielu interesujących substancji. Stwierdzili oni na przykład, że przez destylację surowej smoły można otrzymać frakcje o rozmaitych właściwościach. Z jednych odzyskiwano naftalen, z innych dawało się wyprodukować fenol, zaś najlżejsza i niskowrząca frakcja dawała się zastosować jako rozpuszczalnik i olej oświetleniowy. Frakcja ta stanowiła też zresztą uciążliwe zanieczyszczenie gazu koksowniczego używanego do oświetlenia, zauważalne zwłaszcza gdy doprowadzany gaz był jeszcze ciepły. Wykraplała się na chłodnych kloszach latarń i przemieszana z sadzą zbierała na dnie.
Tam też na lepkie zanieczyszczenie zwrócił uwagę w 1825 roku Michael Faraday, który będąc bardzo praktycznym człowiekiem podjął się jej destylacji, chcąc otrzymać palny olej. Przydatnym produktem okazała się jedna z frakcji, o temperaturze wrzenia 80°C. Była to rzadka, lekko żółtawa ciecz spalająca się bardzo kopcącym płomieniem i będąca dobrym rozpuszczalnikiem. W następnych dekadach nauczono się wyodrębniać ją na duża skalę ze smoły pogazowej, a ze względu na obfite występowanie w benzolu, cieczy absorbowanej z gazu koksowniczego na stałych pochłaniaczach, nazwano ją benzenem.
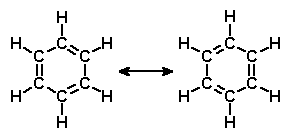
Benzen odegrał dużą rolę w rozwoju chemii organicznej. To od niego pewną grupę niereaktywnych związków, często posiadających charakterystyczny zapach, nazwano związkami aromatycznymi. W tym wczesnym okresie duży problem sprawiało chemikom określenie jego struktury cząsteczkowej. Całkiem niedawno przyjęło się uważać, że pierwiastki składają się z atomów, a związki ze złożeń tych atomów w drobne całostki, nazwane cząsteczkami, o określonej budowie i układzie połączonych atomów. Jedyną informację o przypuszczalnym składzie cząsteczki stanowiły stosunki ilościowe pierwiastków składowych. Wiedząc w jakich ilościach łączą się ze sobą atomy, należało domyśleć się jaką prawdopodobnie tworzyły ze sobą strukturę.
Chemikiem który włożył w tą dziedzinę najwięcej, był opisywany już tutaj
August Friedrich Kekule. On to po raz pierwszy na podstawie swych badań ustalił, że węgiel w związkach organicznych łączy się z maksymalnie czterema innymi atomami. W późniejszym okresie zastanawiając się jak połączyć ze sobą budulcowe atomy, doszedł do wniosku, że atomy węgla w bardziej skomplikowanych związkach muszą łączyć się tworząc łańcuchy. Wedle opowiadanej przezeń po latach anegdoty, myśl tą podsunął mu sen w którym dostrzegł tańczące atomy, które w pewnym momencie zaczęły się bawić w lokomotywę.
Pomysł ten nie dawał się jednak zastosować do niektórych związków, czego przykładem był benzen, złożony z węgla i wodoru w stosunku 1:1, i zawierający najwyraźniej sześć węgli. Rozwiązanie podsunął mu kolejny sen, w którym tańczące atomy utworzyły węża, a ten w pewnym momencie uchwycił swój ogon i w takiej formie wirował mu przed oczami. No tak - załóżmy że atomy są połączone w pierścień i mają wolną możliwość przyłączenia jeszcze tylko po jednym, a skład będzie się zgadzał.
Po upływie kolejnych lat chemicy coraz śmielej poczynali sobie z tworzeniem nowych pochodnych tego związku, aż w roku 1879 słynny chemik
Bayer, założyciel zakładu produkującego między innymi Aspirynę, zauważył bardzo specyficzną reakcję - gdy wytrząsnął benzen ze stężonym kwasem siarkowym i dodał izatyny, żółtopomarańczowej substancji otrzymywanej z indygo, powstawało wyraźne niebieskie zabarwienie, zauważalne nawet przy niewielkich ilościach substancji. Wyglądało zatem na to, że odkryto prostą i szybką reakcję charakterystyczną, pozwalającą wykrywać benzen.
Odkrycie szybko zostało uznane i niektórzy postępowi profesorowie chemii zaczęli uczyć o tej reakcji na uniwersytetach. Jednym z nich był profesor Wiliam Weith wykładający chemię na uniwersytecie w Zurychu. Miał on specjalny lektorat poświęcony związkom aromatycznym, podczas którego pokazywał najbardziej charakterystyczne reakcje. Niestety na początku 1882 roku zmarł, toteż zajęciami podczas wiosennego semestru zajął się jego bliski przyjaciel
Viktor Meyer.
Gdy przygotowywał się do zajęć polecił swojemu asystentowi aby przygotował mu próbkę benzenu. Tylko miał być czysty, tak aby pokaz poszedł bez problemów.
W dniu wykładu asystent dostarczył odpowiednią ilość związku. Meyer omówił historię i strukturę benzenu, po czym przeszedł do omawiania reakcji. Można wyobrazić sobie jak mówi studentom, że gdy teraz wytrząsie benzen ze stężonym kwasem i doda izatyny, to zobaczymy piękny niebieski kolor. Następnie tak jak mówił wytrząsa w próbówce benzen i stężony kwas, dodaje izatynę i... nic się nie dzieje. Powtarza reakcję, bo może coś akurat źle zrobił, ale nic nie pomaga. No cóż, tak się czasem zdarza, powtórzymy na następnych zajęciach.
Po skończonym wykładzie zwrócił się zatem do asystenta z delikatnym zapytaniem, co on u licha mu na te zajęcia przygotował. Bo jeśli nie szyny i nie izatyna, to benzen był zły. Asystent, znany później Traugott Sandmeyer bronił się że ależ skąd, przygotował benzen czysty, jak profesor chciał, wszystko wedle przepisu z dekarboksylacji kwasu benzoesowego bo tylko wtedy dawało się otrzymać zupełnie czysty. To już było zastanawiające. Jeszcze tego samego dnia Meyer wziął komercyjnie dostępny benzen otrzymywany z powęglowego benzolu, wytrząsnął z kwasem, dodał izatyny i otrzymał zgodnie z opisem Bayera piękny niebieski barwnik, znany jako
indofenina.
Nie wiedząc co z tym faktem począć, wziął większą ilość komercyjnego benzenu, wytrząsnął z kwasem, oddzieloną warstwę kwasową zobojętnił stwierdzając, że wydzieliła mu się rzadka, lekko żółtawa ciecz o charakterystycznym zapachu, która wydawała się identyczna z benzenem. Meyer sądził zatem, że benzen występuje w dwóch formach, jednej mało aktywnej i drugiej "zaktywizowanej" i wchodzącej w reakcję barwną. Powtórzenie reakcji z otrzymaną cieczą pozwoliło mu na wytworzenie większej ilości niebieskiego barwnika, który wysłał do zbadania Bayerowi. Ten orzekł, że faktycznie jest to indofenina, ale zarazem w analizie elementarnej wyszło mu, że związek zawiera siarkę, której nie było w izatynie. Dalsze testy "aktywizowanego benzenu" pokazały, że musi być to substancja różna od benzenu. W odróżnieniu od niego nie krystalizowała w lodzie, i wrzała w temperaturze 84 stopni, w porównaniu z 80 stopni dla benzenu zupełnie czystego. Wreszcie analiza chemiczna wykazała, że jest to związek zawierający jeden atom siarki, cztery atomy węgla i cztery wodoru.
I tak Meyer odkrył Tiofen.
Odkrycie tiofenu zelektryzowało ówczesnych chemików. Okazało się że przez kilka dekad nie zauważyli, że benzen ze smoły węglowej jest mieszanką dwóch związków, przy czym ten drugi, tiofen, stanowił w niektórych partiach do 10%
Tiofen należy do grupy pięciokątnych związków aromatycznych, w których aromatyczność nadaje im zdelokalizowany układ sześciu elektronów - dwóch pochodzących z wiązań podwójnych na części węglowej i jednej wolnej pary pożyczonej z heteroatomu. Pełnowęglowy odpowiednik czyli cyklopentadien nie jest aromatyczny, a dodatkowo efekty antyaromatyczne tylko zmniejszają jego trwałość. Dążąc do utrwalenia chętnie odszczepia jeden wodór tworząc karboanion cyklopentadienylowy który już jest aromatyczny.
Podstawienie jednego węgla w tym układzie heteroatomem posiadającym wolną parę elektronową tworzy aromatyczną cząsteczkę obojętną. Gdy tym atomem jest tlen, otrzymujemy
furan, gdy azot jest to
pirol. Udało się także otrzymać analogiczne cząsteczki z niektórymi metalami i półmetalami, takie jak
silol z krzemem,
arsol z arsenem,
stannol z cyną a nawet
tytanol z tytanem. Zachowują one częściową aromatyczność, ale znacznie osłabioną.
Dziś możemy już odpowiedzieć na pytanie co takiego zachodziło w próbówce Meyera i co właściwie wykrywała reakcja. Tiofen w odróżnieniu od benzenu jest bardziej reaktywny. Tyle samo bo sześć elektronów stłoczonych jest jednak na mniejszym bo pięcioatomowym pierścieniu. Większe zagęszczenie ładunku (oraz karboanionowe struktury mezomeryczne) powoduje, że chętniej reaguje z czynnikami elektrofilowymi. Takim czynnikiem może być też proton uwalniany przez odpowiednio silny kwas.
Podczas wytrząsania benzolu ze stężonym kwasem, tiofen ulegał protonowaniu i w formie jonowej przechodził do warstwy kwasowej. Dalsza reakcja z izatyną jest dość skomplikowana i nie zupełnie rozgryziona, zaczyna się prawdopodobnie od sprotonowania izatyny i wytworzenia formy z ładunkiem dodatnim, która jako elektrofil atakuje cząsteczkę tiofenu. Powstające połączenie dimeryzuje i ulega przegrupowaniu tworząc niebieski barwnik:
Indofenina występuje w kilku izomerach różniących się konformacją trans/cis na wiązaniach podwójnych, w zasadzie więc powstaje mieszanina izomerów. Reakcja ma dziś jeszcze zastosowanie do oznaczania niektórych mało podstawionych pochodnych tiofenu.
Jakie zastosowania ma tiofen?
Jednym które samo się narzuca jest produkcja barwników. Chętnie jest też używany w syntezach nowych leków. Może zastępować pierścień benzenowy bez utraty właściwości leku, a dzięki łatwiejszemu podstawieniu łatwiej jest wytworzyć różnorodne pochodne.
Najciekawszym zastosowaniem jest jednak wytwarzanie
politiofenu, polimeru mogącego przewodzić prąd elektryczny.
Spolimeryzowany tiofen po utlenieniu staje się przewodnikiem typu metalicznego. Utleniony tylko częściowo stanowi natomiast organiczny półprzewodnik. Możliwe jest więc wytworzenie na przykład przezroczystej folii przewodzącej prąd, co powinno znaleźć zastosowanie w ogniwach słonecznych. Szersze zastosowanie znalazła dobrze rozpuszczalna pochodna poli(etylenodioksytiofenu) (PEDOT-PSS), która dzięki przewodnictwu jest używana w powłokach antystatycznych, nie pozwalających na elektryzowanie się powierzchni.
Sam poli(etylenodioksytiofen) jest słabo rozpuszczalny w rozpuszczalnikach organicznych. Folie i przewody wytworzone z tego materiału są używane w elastycznych wyświetlaczach OLED.
------------
*
H. D. Hartough, The Chemistry of Heterocyclic Compounds, Thiophene and Its Derivatives,
* https://en.wikipedia.org/wiki/Thiophene
* https://en.wikipedia.org/wiki/Polythiophene